

A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction
- PMID: 31801360
- PMCID: PMC7147986
- DOI: 10.1161/CIRCULATIONAHA.119.042178
A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction
Abstract
Background: Heart failure is a leading cause of death worldwide. Cyclic nucleotide phosphodiesterases (PDEs), through degradation of cyclic nucleotides, play critical roles in cardiovascular biology and disease. Our preliminary screening studies have revealed PDE10A upregulation in the diseased heart. However, the roles of PDE10A in cardiovascular biology and disease are largely uncharacterized. The current study is aimed to investigate the regulation and function of PDE10A in cardiac cells and in the progression of cardiac remodeling and dysfunction.
Methods: We used isolated adult mouse cardiac myocytes and fibroblasts, as well as preclinical mouse models of hypertrophy and heart failure. The PDE10A selective inhibitor TP-10, and global PDE10A knock out mice were used.
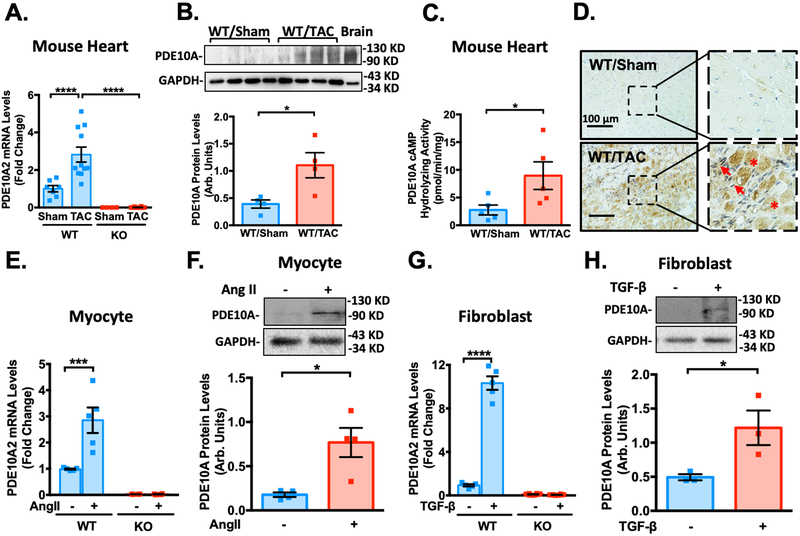
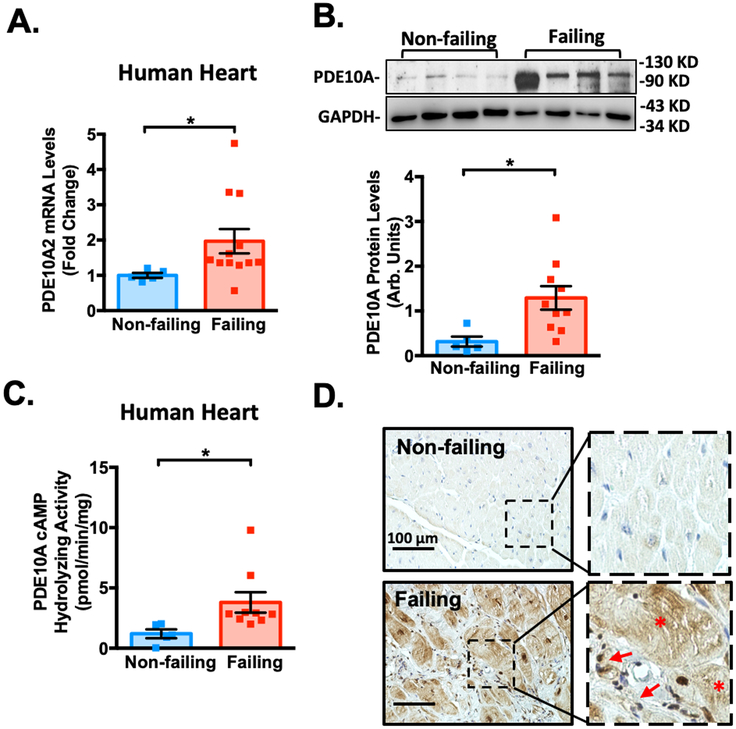
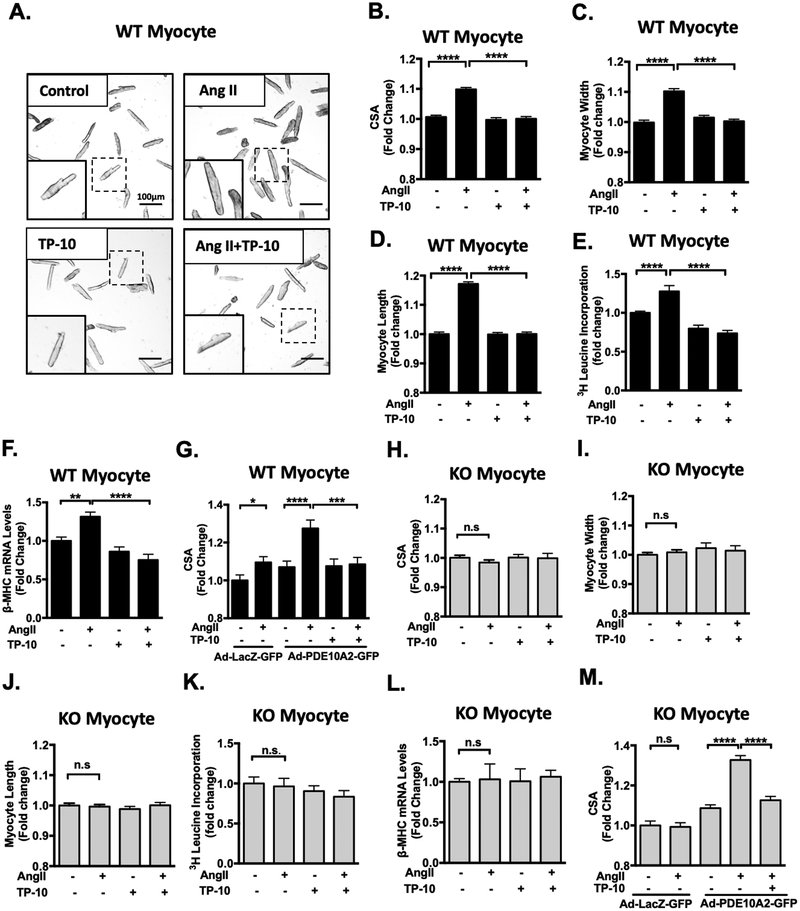
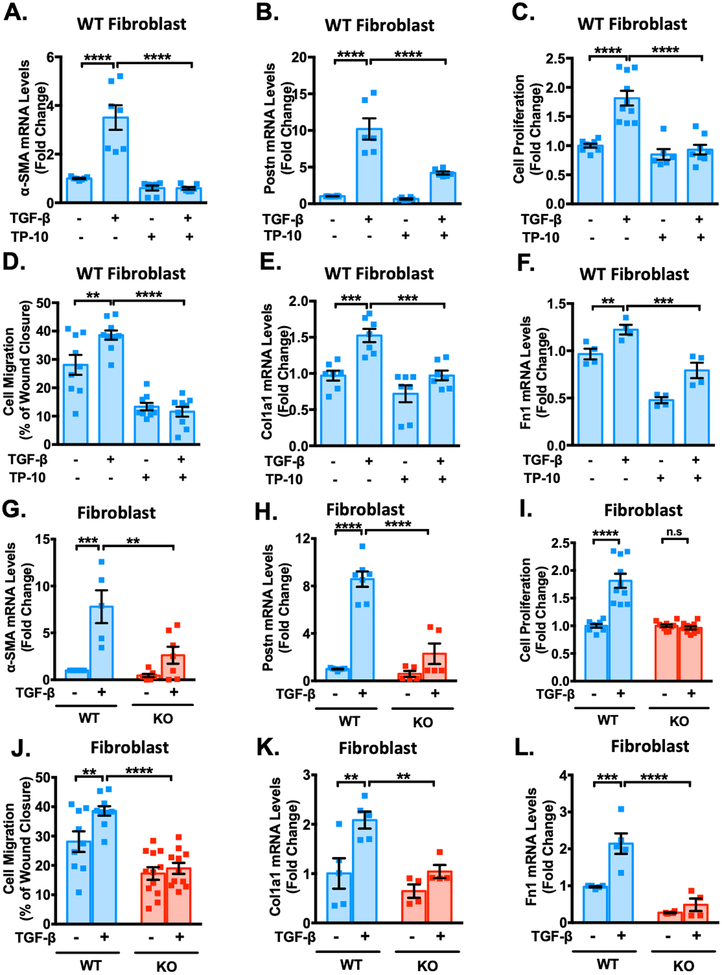
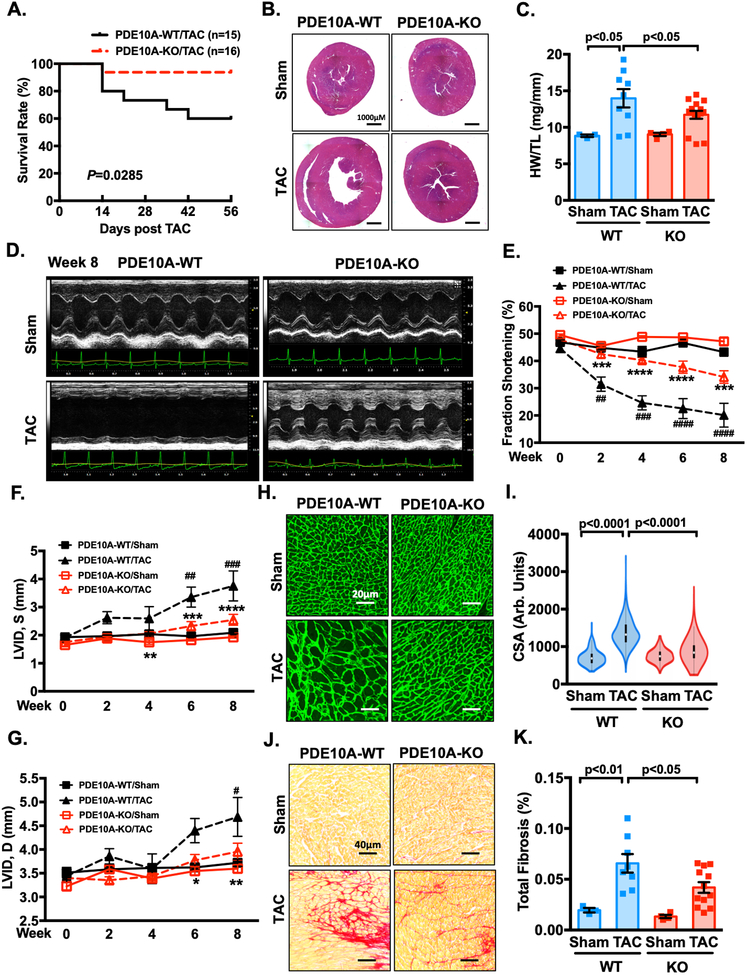
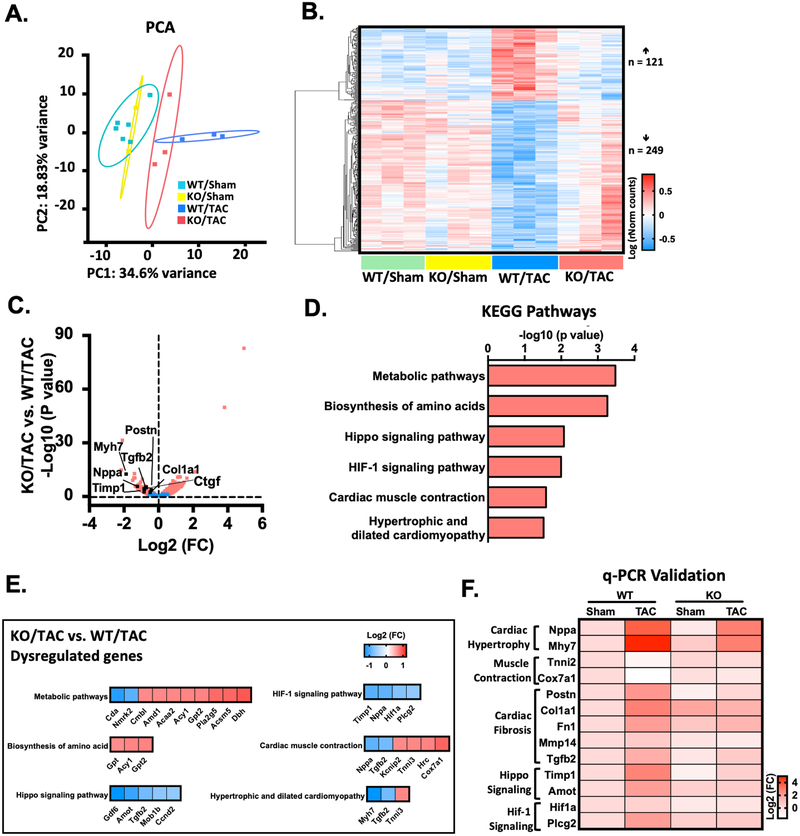
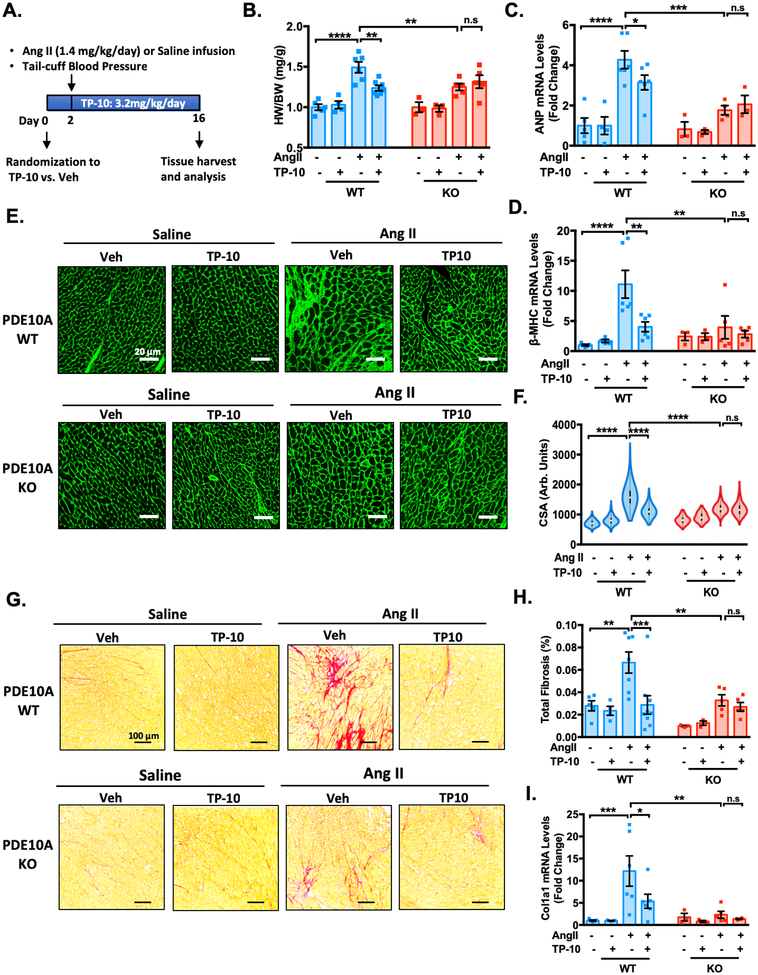
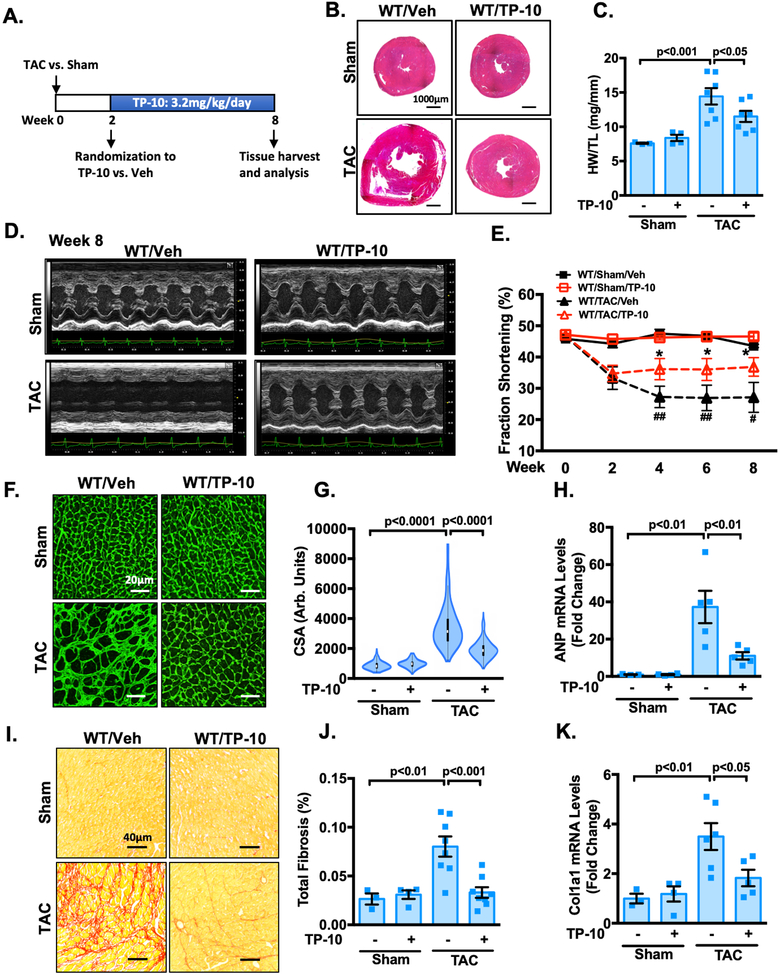
Results: We found that PDE10A expression remains relatively low in normal and exercised heart tissues. However, PDE10A is significantly upregulated in mouse and human failing hearts. In vitro, PDE10A deficiency or inhibiting PDE10A with selective inhibitor TP-10, attenuated cardiac myocyte pathological hypertrophy induced by Angiotensin II, phenylephrine, and isoproterenol, but did not affect cardiac myocyte physiological hypertrophy induced by IGF-1 (insulin-like growth factor 1). TP-10 also reduced TGF-β (transforming growth factor-β)-stimulated cardiac fibroblast activation, proliferation, migration and extracellular matrix synthesis. TP-10 treatment elevated both cAMP and cGMP levels in cardiac myocytes and cardiac fibroblasts, consistent with PDE10A as a cAMP/cGMP dual-specific PDE. In vivo, global PDE10A deficiency significantly attenuated myocardial hypertrophy, cardiac fibrosis, and dysfunction induced by chronic pressure overload via transverse aorta constriction or chronic neurohormonal stimulation via Angiotensin II infusion. Importantly, we demonstrated that the pharmacological effect of TP-10 is specifically through PDE10A inhibition. In addition, TP-10 is able to reverse pre-established cardiac hypertrophy and dysfunction. RNA-Sequencing and bioinformatics analysis further identified a PDE10A-regualted transcriptome involved in cardiac hypertrophy, fibrosis, and cardiomyopathy.
Conclusions: Taken together, our study elucidates a novel role for PDE10A in the regulation of pathological cardiac remodeling and development of heart failure. Given that PDE10A has been proven to be a safe drug target, PDE10A inhibition may represent a novel therapeutic strategy for preventing and treating cardiac diseases associated with cardiac remodeling.
Keywords: cardiac hypertrophy; cyclic nucleotide phosphodiesterases.
Figures
Comment in
-
Response by Chen and Yan to Letter Regarding Article, "A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction".Circulation. 2020 Jul 21;142(3):e36-e37. doi: 10.1161/CIRCULATIONAHA.120.048259. Epub 2020 Jul 20. Circulation. 2020. PMID: 32687445 Free PMC article. No abstract available.
-
Letter by Wang et al Regarding Article, "A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction".Circulation. 2020 Jul 21;142(3):e34-e35. doi: 10.1161/CIRCULATIONAHA.120.046614. Epub 2020 Jul 20. Circulation. 2020. PMID: 32687447 No abstract available.
Similar articles
-
PDE10A Inactivation Prevents Doxorubicin-Induced Cardiotoxicity and Tumor Growth.Circ Res. 2023 Jul 7;133(2):138-157. doi: 10.1161/CIRCRESAHA.122.322264. Epub 2023 May 26. Circ Res. 2023. PMID: 37232184 Free PMC article.
-
Vinpocetine Attenuates Pathological Cardiac Remodeling by Inhibiting Cardiac Hypertrophy and Fibrosis.Cardiovasc Drugs Ther. 2017 Apr;31(2):157-166. doi: 10.1007/s10557-017-6719-0. Cardiovasc Drugs Ther. 2017. PMID: 28321644 Free PMC article.
-
Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy.Circ Res. 2009 Nov 6;105(10):956-64. doi: 10.1161/CIRCRESAHA.109.198515. Epub 2009 Sep 24. Circ Res. 2009. PMID: 19797176 Free PMC article.
-
Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiac hypertrophy and heart failure.Nat Rev Cardiol. 2023 Feb;20(2):90-108. doi: 10.1038/s41569-022-00756-z. Epub 2022 Sep 1. Nat Rev Cardiol. 2023. PMID: 36050457 Review.
-
Cardiac Cyclic Nucleotide Phosphodiesterases: Roles and Therapeutic Potential in Heart Failure.Cardiovasc Drugs Ther. 2020 Jun;34(3):401-417. doi: 10.1007/s10557-020-06959-1. Cardiovasc Drugs Ther. 2020. PMID: 32172427 Free PMC article. Review.
Cited by
-
Inhibitory effect of (pro)renin receptor decoy inhibitor PRO20 on endoplasmic reticulum stress during cardiac remodeling.Front Pharmacol. 2022 Aug 12;13:940365. doi: 10.3389/fphar.2022.940365. eCollection 2022. Front Pharmacol. 2022. PMID: 36034809 Free PMC article.
-
SIRT6 Mitigates Heart Failure With Preserved Ejection Fraction in Diabetes.Circ Res. 2022 Nov 11;131(11):926-943. doi: 10.1161/CIRCRESAHA.121.318988. Epub 2022 Oct 24. Circ Res. 2022. PMID: 36278398 Free PMC article.
-
Epigenome-wide DNA methylation profiling in comparison between pathological and physiological hypertrophy of human cardiomyocytes.Front Genet. 2023 Sep 27;14:1264382. doi: 10.3389/fgene.2023.1264382. eCollection 2023. Front Genet. 2023. PMID: 37829282 Free PMC article.
-
Cyclic GMP modulating drugs in cardiovascular diseases: mechanism-based network pharmacology.Cardiovasc Res. 2022 Jul 20;118(9):2085-2102. doi: 10.1093/cvr/cvab240. Cardiovasc Res. 2022. PMID: 34270705 Free PMC article. Review.
-
PDE10A Inactivation Prevents Doxorubicin-Induced Cardiotoxicity and Tumor Growth.Circ Res. 2023 Jul 7;133(2):138-157. doi: 10.1161/CIRCRESAHA.122.322264. Epub 2023 May 26. Circ Res. 2023. PMID: 37232184 Free PMC article.
References
-
- Frey N and Olson EN. Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol. 2003;65:45–79. - PubMed
-
- Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P, American Heart Association Council on E, Prevention Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137:e67–e492. - PubMed
-
- Hill JA and Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. - PubMed
-
- Porter KE and Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009;123:255–278. - PubMed
-
- Guellich A, Mehel H and Fischmeister R. Cyclic AMP synthesis and hydrolysis in the normal and failing heart. Pflugers Arch. 2014;466:1163–1175. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
