

Risk-adapted therapy and biological heterogeneity in pineoblastoma: integrated clinico-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials
- PMID: 31802236
- PMCID: PMC7065912
- DOI: 10.1007/s00401-019-02106-9
Risk-adapted therapy and biological heterogeneity in pineoblastoma: integrated clinico-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials
Erratum in
-
Correction to: Risk-adapted therapy and biological heterogeneity in pineoblastoma: integrated clinico-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials.Acta Neuropathol. 2020 Feb;139(2):273-275. doi: 10.1007/s00401-019-02115-8. Acta Neuropathol. 2020. PMID: 31865440
Abstract
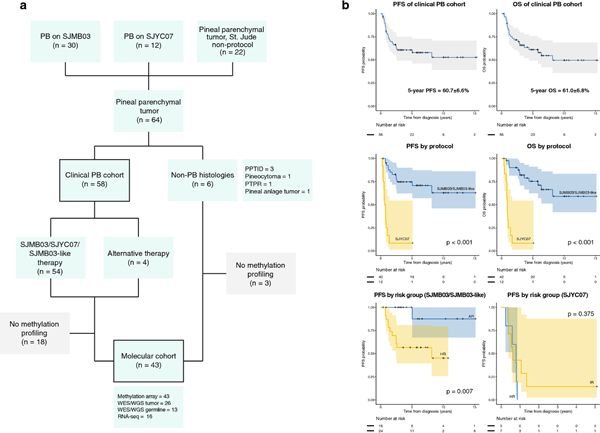
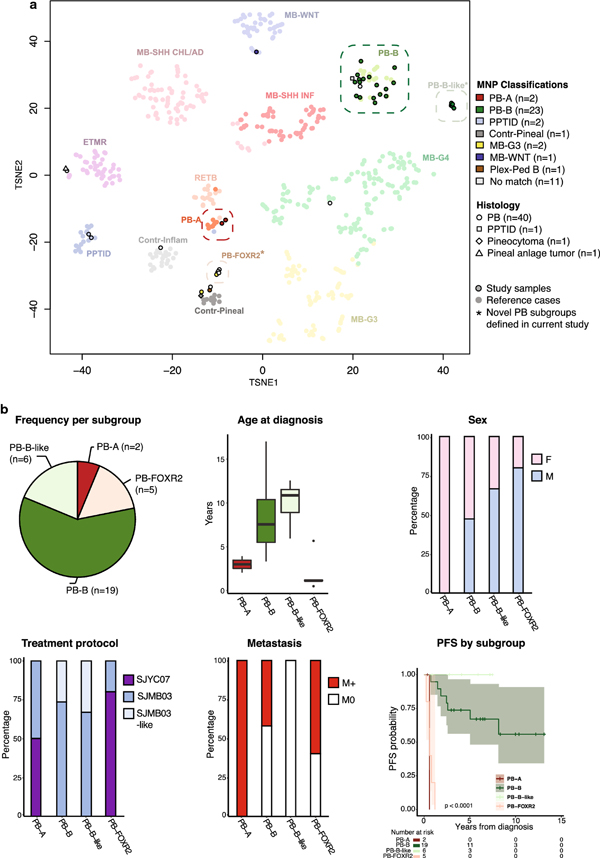
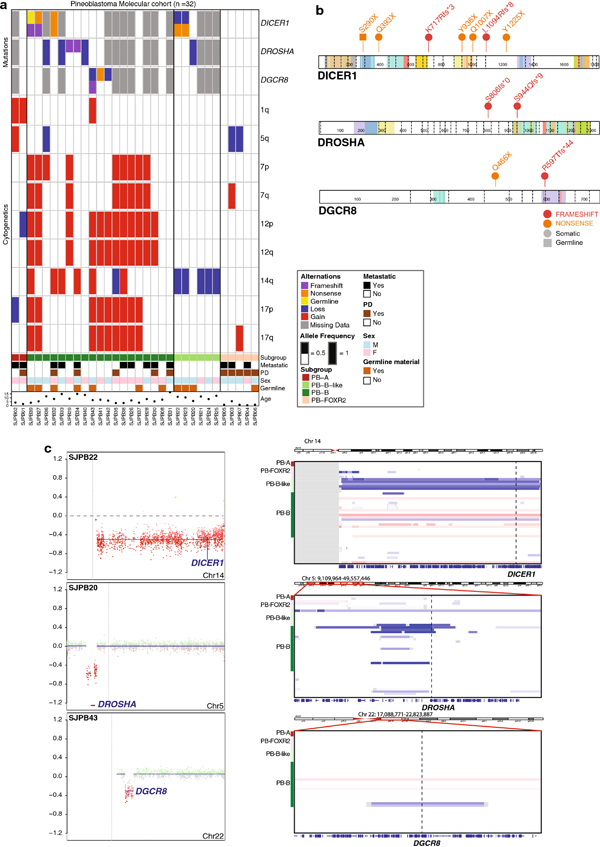
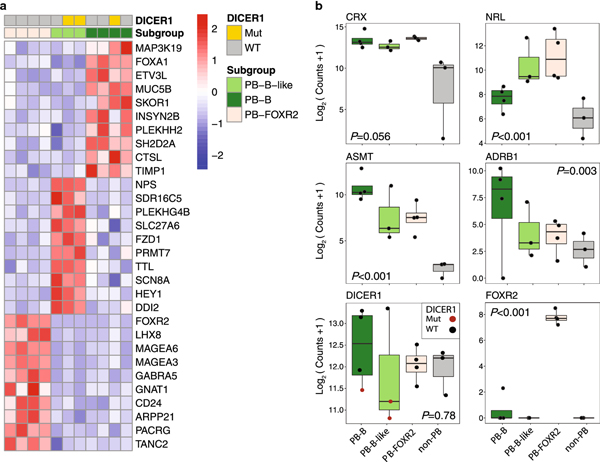
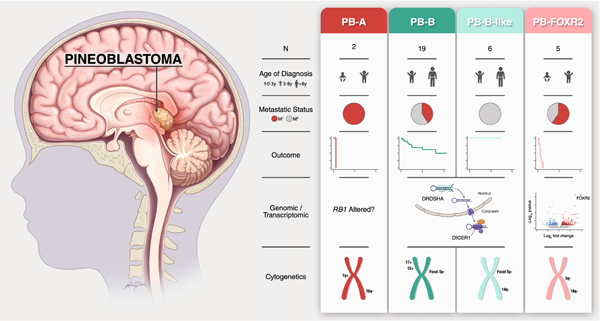
Pineoblastoma is a rare embryonal tumor of childhood that is conventionally treated with high-dose craniospinal irradiation (CSI). Multi-dimensional molecular evaluation of pineoblastoma and associated intertumoral heterogeneity is lacking. Herein, we report outcomes and molecular features of children with pineoblastoma from two multi-center, risk-adapted trials (SJMB03 for patients ≥ 3 years; SJYC07 for patients < 3 years) complemented by a non-protocol institutional cohort. The clinical cohort consisted of 58 patients with histologically diagnosed pineoblastoma (SJMB03 = 30, SJYC07 = 12, non-protocol = 16, including 12 managed with SJMB03-like therapy). The SJMB03 protocol comprised risk-adapted CSI (average-risk = 23.4 Gy, high-risk = 36 Gy) with radiation boost to the primary site and adjuvant chemotherapy. The SJYC07 protocol consisted of induction chemotherapy, consolidation with focal radiation (intermediate-risk) or chemotherapy (high-risk), and metronomic maintenance therapy. The molecular cohort comprised 43 pineal parenchymal tumors profiled by DNA methylation array (n = 43), whole-exome sequencing (n = 26), and RNA-sequencing (n = 16). Respective 5-year progression-free survival rates for patients with average-risk or high-risk disease on SJMB03 or SJMB03-like therapy were 100% and 56.5 ± 10.3% (P = 0.007); respective 2-year progression-free survival rates for those with intermediate-risk or high-risk disease on SJYC07 were 14.3 ± 13.2% and 0% (P = 0.375). Of patients with average-risk disease treated with SJMB03/SJMB03-like therapy, 17/18 survived without progression. DNA-methylation analysis revealed four clinically relevant pineoblastoma subgroups: PB-A, PB-B, PB-B-like, and PB-FOXR2. Pineoblastoma subgroups differed in age at diagnosis, propensity for metastasis, cytogenetics, and clinical outcomes. Alterations in the miRNA-processing pathway genes DICER1, DROSHA, and DGCR8 were recurrent and mutually exclusive in PB-B and PB-B-like subgroups; PB-FOXR2 samples universally overexpressed the FOXR2 proto-oncogene. Our findings suggest superior outcome amongst older children with average-risk pineoblastoma treated with reduced-dose CSI. The identification of biologically and clinically distinct pineoblastoma subgroups warrants consideration of future molecularly-driven treatment protocols for this rare pediatric brain tumor entity.
Keywords: Clinical trial; DICER1; FOXR2; MicroRNA processing; Molecular subgroups; Pineoblastoma.
Conflict of interest statement
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The authors disclose no conflict of interest.
Figures
References
-
- Chintagumpala M, Hassall T, Palmer S, Ashley D, Wallace D, Kasow K, Merchant TE, Krasin MJ, Dauser R, Boop F, Krance R, Woo S, Cheuk R, Lau C, Gilbertson R, Gajjar A (2009) A pilot study of risk-adapted radiotherapy and chemotherapy in patients with supratentorial PNET. Neuro-oncol 11:33–40. doi:10.1215/15228517-2008-079 - DOI - PMC - PubMed
-
- Cho Y-J, Tsherniak A, Tamayo P, Santagata S, Ligon A, Greulich H, Berhoukim R, Amani V, Goumnerova L, Eberhart CG, Lau CC, Olson JM, Gilbertson RJ, Gajjar A, Delattre O, Kool M, Ligon K, Meyerson M, Mesirov JP, Pomeroy SL (2011) Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. J Clin Oncol 29:1424–1430. doi:10.1200/JCO.2010.28.5148 - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
