

DLC1 SAM domain-binding peptides inhibit cancer cell growth and migration by inactivating RhoA
- PMID: 31806702
- PMCID: PMC6956525
- DOI: 10.1074/jbc.RA119.011929
DLC1 SAM domain-binding peptides inhibit cancer cell growth and migration by inactivating RhoA
Abstract
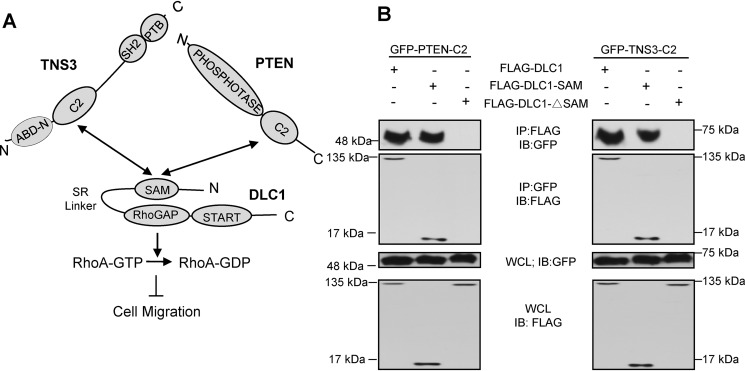
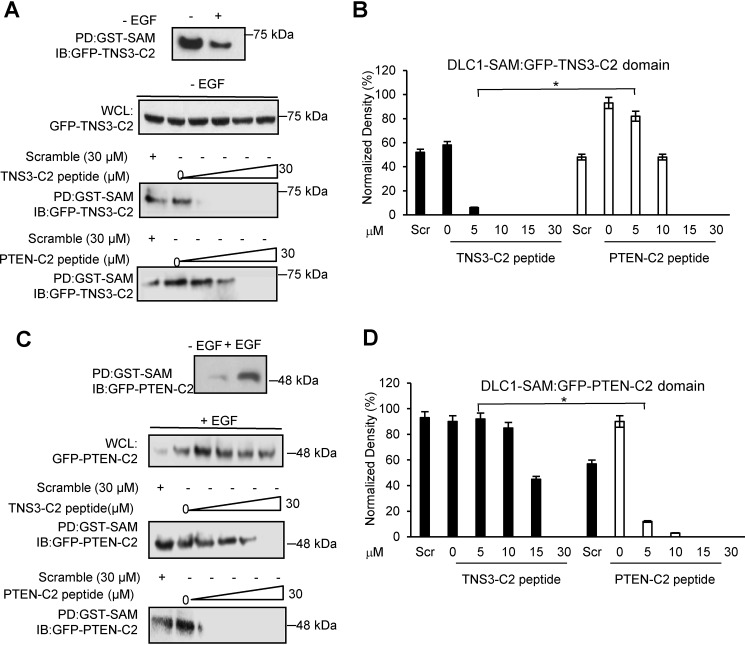
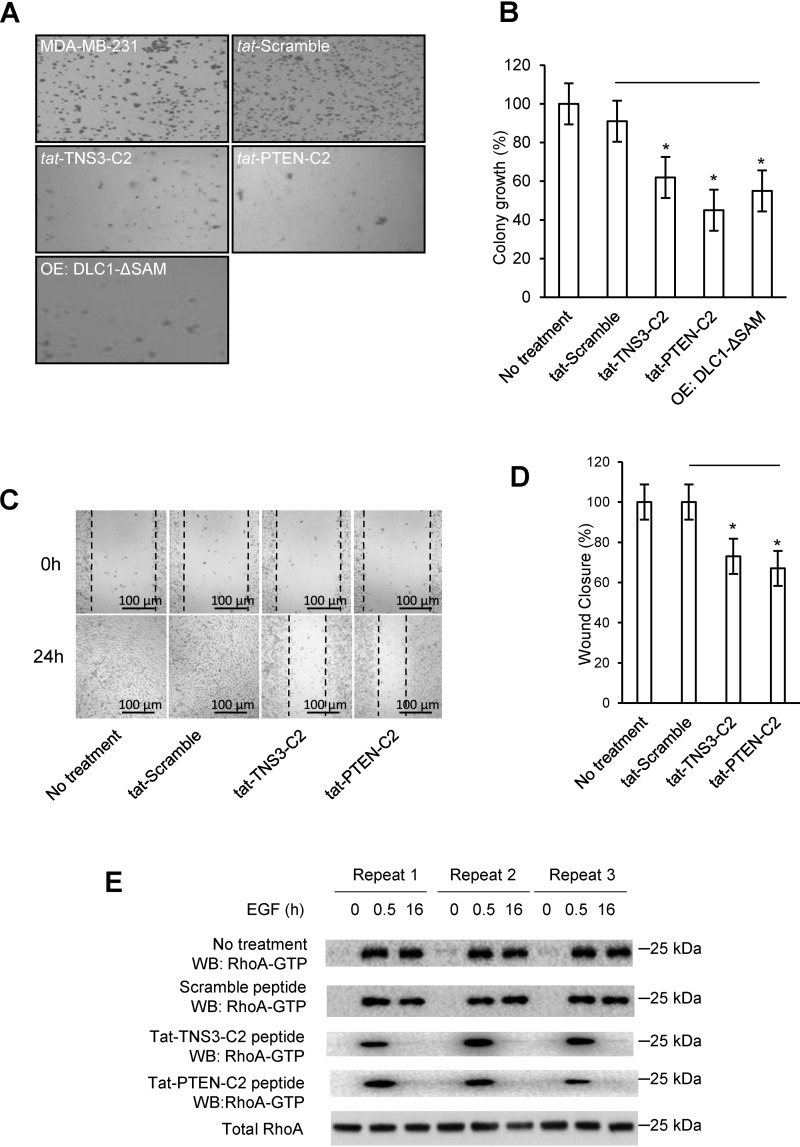
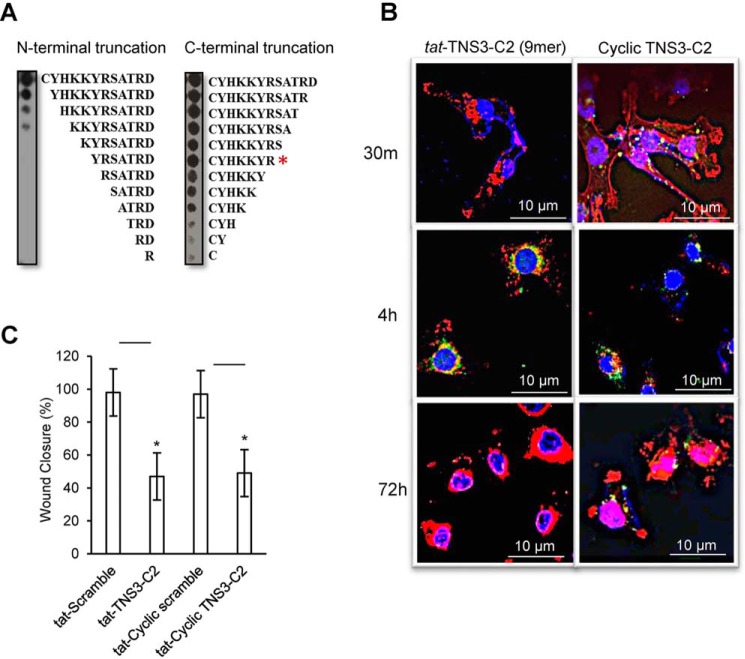
Deleted-in-liver cancer 1 (DLC1) exerts its tumor suppressive function mainly through the Rho-GTPase-activating protein (RhoGAP) domain. When activated, the domain promotes the hydrolysis of RhoA-GTP, leading to reduced cell migration. DLC1 is kept in an inactive state by an intramolecular interaction between its RhoGAP domain and the DLC1 sterile α motif (SAM) domain. We have shown previously that this autoinhibited state of DLC1 may be alleviated by tensin-3 (TNS3) or PTEN. We show here that the TNS3/PTEN-DLC1 interactions are mediated by the C2 domains of the former and the SAM domain of the latter. Intriguingly, the DLC1 SAM domain was capable of binding to specific peptide motifs within the C2 domains. Indeed, peptides containing the binding motifs were highly effective in blocking the C2-SAM domain-domain interaction. Importantly, when fused to the tat protein-transduction sequence and subsequently introduced into cells, the C2 peptides potently promoted the RhoGAP function in DLC1, leading to decreased RhoA activation and reduced tumor cell growth in soft agar and migration in response to growth factor stimulation. To facilitate the development of the C2 peptides as potential therapeutic agents, we created a cyclic version of the TNS3 C2 domain-derived peptide and showed that this peptide readily entered the MDA-MB-231 breast cancer cells and effectively inhibited their migration. Our work shows, for the first time, that the SAM domain is a peptide-binding module and establishes the framework on which to explore DLC1 SAM domain-binding peptides as potential therapeutic agents for cancer treatment.
Keywords: C2 domain; Ras homolog gene family, member A (RhoA); RhoGAP; SAM domain; breast cancer; cell migration; cell proliferation; deleted in liver cancer 1 (DLC1).
© 2020 Joshi et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures


References
-
- Xue W., Krasnitz A., Lucito R., Sordella R., Vanaelst L., Cordon-Cardo C., Singer S., Kuehnel F., Wigler M., Powers S., Zender L., and Lowe S. W. (2008) DLC1 is a chromosome 8p tumor suppressor whose loss promotes hepatocellular carcinoma. Genes Dev. 22, 1439–1444 10.1101/gad.1672608 - DOI - PMC - PubMed
-
- Zhou X., Zimonjic D. B., Park S. W., Yang X. Y., Durkin M. E., and Popescu N. C. (2008) DLC1 suppresses distant dissemination of human hepatocellular carcinoma cells in nude mice through reduction of RhoA GTPase activity, actin cytoskeletal disruption and down-regulation of genes involved in metastasis. Int. J. Oncol. 32, 1285–1291 - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
