

Monocytes prime autoreactive T cells after myocardial infarction
- PMID: 31809213
- PMCID: PMC6985803
- DOI: 10.1152/ajpheart.00595.2019
Monocytes prime autoreactive T cells after myocardial infarction
Abstract
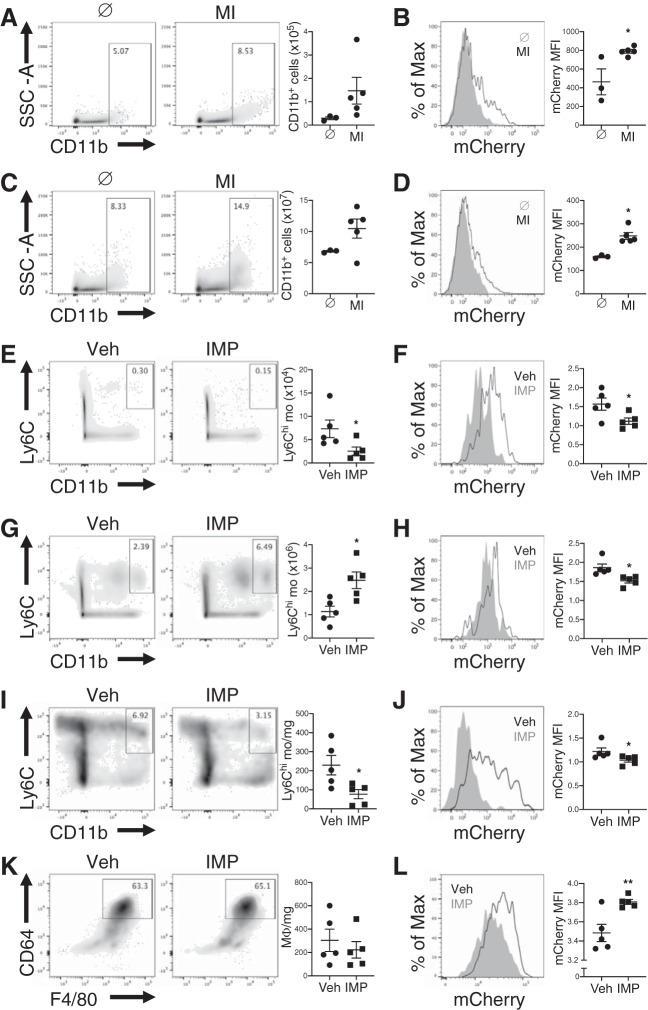
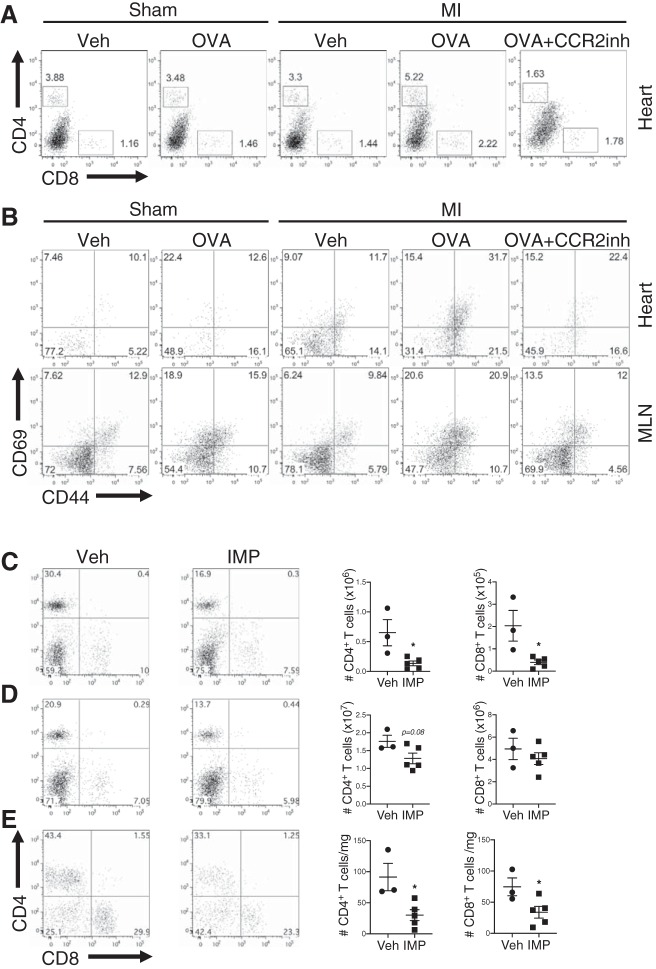
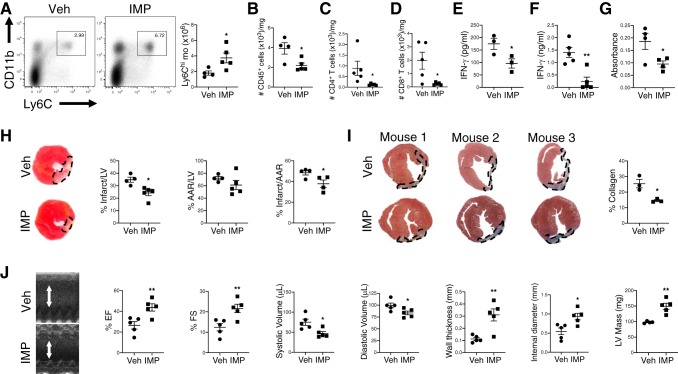
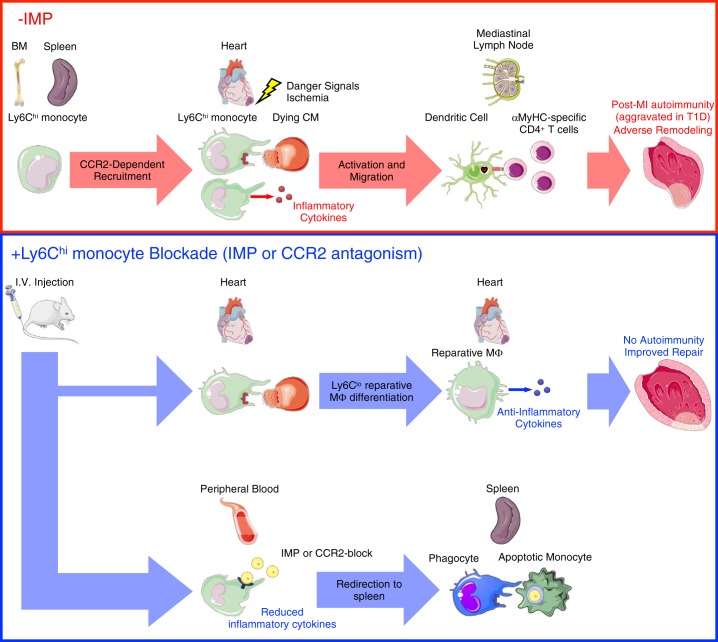
In humans, loss of central tolerance for the cardiac self-antigen α-myosin heavy chain (α-MHC) leads to circulation of cardiac autoreactive T cells and renders the heart susceptible to autoimmune attack after acute myocardial infarction (MI). MI triggers profound tissue damage, releasing danger signals and self-antigen by necrotic cardiomyocytes, which lead to recruitment of inflammatory monocytes. We hypothesized that excessive inflammation by monocytes contributes to the initiation of adaptive immune responses to cardiac self-antigen. Using an experimental model of MI in α-MHC-mCherry reporter mice, which specifically express mCherry in cardiomyocytes, we detected α-MHC antigen in myeloid cells in the heart-draining mediastinal lymph node (MLN) 7 days after MI. To test whether monocytes were required for cardiac self-antigen trafficking to the MLN, we blocked monocyte recruitment with a C-C motif chemokine receptor type 2 (CCR2) antagonist or immune modifying nanoparticles (IMP). Blockade of monocyte recruitment reduced α-MHC antigen detection in the MLN after MI. Intramyocardial injection of the model antigen ovalbumin into OT-II transgenic mice demonstrated the requirement for monocytes in antigen trafficking and T-cell activation in the MLN. Finally, in nonobese diabetic mice, which are prone to postinfarction autoimmunity, blockade of monocyte recruitment reduced α-MHC-specific responses leading to improved tissue repair and ventricular function 28 days after MI. Taken together, these data support a role for monocytes in the onset of pathological cardiac autoimmunity following MI and suggest that therapeutic targeting of monocytes may mitigate postinfarction autoimmunity in humans.NEW & NOTEWORTHY Our study newly identifies a role for inflammatory monocytes in priming an autoimmune T-cell response after myocardial infarction. Select inhibition of monocyte recruitment to the infarct prevents trafficking of cardiac self-antigen and activation of cardiac myosin reactive T cells in the heart-draining lymph node. Therapeutic targeting of inflammatory monocytes may limit autoimmune responses to improve cardiac remodeling and preserve left ventricular function after myocardial infarction.
Keywords: autoimmunity; monocytes; myocardial infarction.
Conflict of interest statement
S. Miller is a cofounder, member of the scientific advisory board, and consultant of Cour Pharmaceutical Development, Co., which is partnering with Takeda Pharmaceutical, Co., for clinical translation of the immune modifying nanoparticle tolerance technology.
Figures
Similar articles
-
C-X-C Motif Chemokine Receptor 4 Blockade Promotes Tissue Repair After Myocardial Infarction by Enhancing Regulatory T Cell Mobilization and Immune-Regulatory Function.Circulation. 2019 Apr 9;139(15):1798-1812. doi: 10.1161/CIRCULATIONAHA.118.036053. Circulation. 2019. PMID: 30696265 Free PMC article.
-
Ly6Chigh Monocytes Oscillate in the Heart During Homeostasis and After Myocardial Infarction-Brief Report.Arterioscler Thromb Vasc Biol. 2017 Sep;37(9):1640-1645. doi: 10.1161/ATVBAHA.117.309259. Epub 2017 Jun 29. Arterioscler Thromb Vasc Biol. 2017. PMID: 28663258
-
The chemokine decoy receptor D6 prevents excessive inflammation and adverse ventricular remodeling after myocardial infarction.Arterioscler Thromb Vasc Biol. 2012 Sep;32(9):2206-13. doi: 10.1161/ATVBAHA.112.254409. Epub 2012 Jul 12. Arterioscler Thromb Vasc Biol. 2012. PMID: 22796582
-
Role of the immune system in cardiac tissue damage and repair following myocardial infarction.Inflamm Res. 2017 Sep;66(9):739-751. doi: 10.1007/s00011-017-1060-4. Epub 2017 Jun 9. Inflamm Res. 2017. PMID: 28600668 Review.
-
The Role of Immune Cells in Cardiac Remodeling After Myocardial Infarction.J Cardiovasc Pharmacol. 2020 Oct;76(4):407-413. doi: 10.1097/FJC.0000000000000876. J Cardiovasc Pharmacol. 2020. PMID: 33027195 Free PMC article. Review.
Cited by
-
PLG nanoparticles target fibroblasts and MARCO+ monocytes to reverse multiorgan fibrosis.JCI Insight. 2022 Mar 8;7(5):e151037. doi: 10.1172/jci.insight.151037. JCI Insight. 2022. PMID: 35104243 Free PMC article.
-
Guidelines for in vivo mouse models of myocardial infarction.Am J Physiol Heart Circ Physiol. 2021 Dec 1;321(6):H1056-H1073. doi: 10.1152/ajpheart.00459.2021. Epub 2021 Oct 8. Am J Physiol Heart Circ Physiol. 2021. PMID: 34623181 Free PMC article. Review.
-
Role of CCR2-Positive Macrophages in Pathological Ventricular Remodelling.Biomedicines. 2022 Mar 12;10(3):661. doi: 10.3390/biomedicines10030661. Biomedicines. 2022. PMID: 35327464 Free PMC article. Review.
-
Inhibition of SphK1/S1P Signaling Pathway Alleviates Fibrosis and Inflammation of Rat Myocardium after Myocardial Infarction.Comput Math Methods Med. 2022 Jul 13;2022:5985375. doi: 10.1155/2022/5985375. eCollection 2022. Comput Math Methods Med. 2022. Retraction in: Comput Math Methods Med. 2023 Jul 12;2023:9797315. doi: 10.1155/2023/9797315. PMID: 35872958 Free PMC article. Retracted.
-
Antigen presentation plays positive roles in the regenerative response to cardiac injury in zebrafish.Nat Commun. 2024 Apr 29;15(1):3637. doi: 10.1038/s41467-024-47430-1. Nat Commun. 2024. PMID: 38684665 Free PMC article.
References
-
- DeBerge M, Yeap XY, Dehn S, Zhang S, Grigoryeva L, Misener S, Procissi D, Zhou X, Lee DC, Muller WA, Luo X, Rothlin C, Tabas I, Thorp EB. MerTK cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ Res 121: 930–940, 2017. doi:10.1161/CIRCRESAHA.117.311327. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials