

Multiscale analysis for patterns of Zika virus genotype emergence, spread, and consequence
- PMID: 31809512
- PMCID: PMC6897431
- DOI: 10.1371/journal.pone.0225699
Multiscale analysis for patterns of Zika virus genotype emergence, spread, and consequence
Abstract
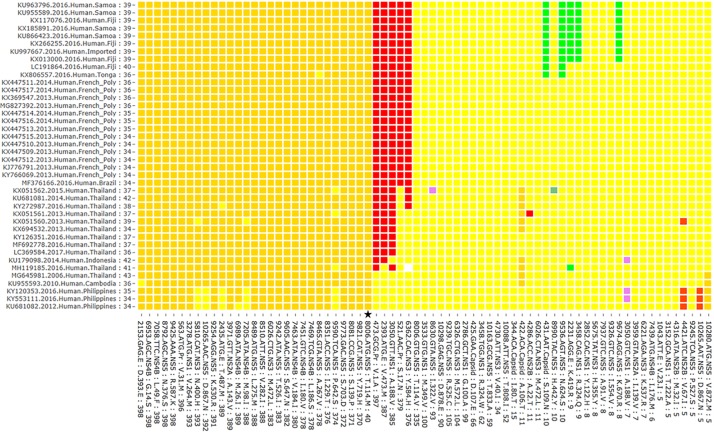
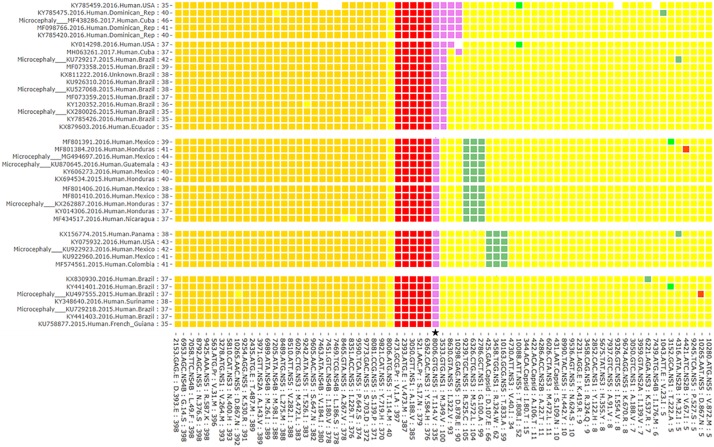
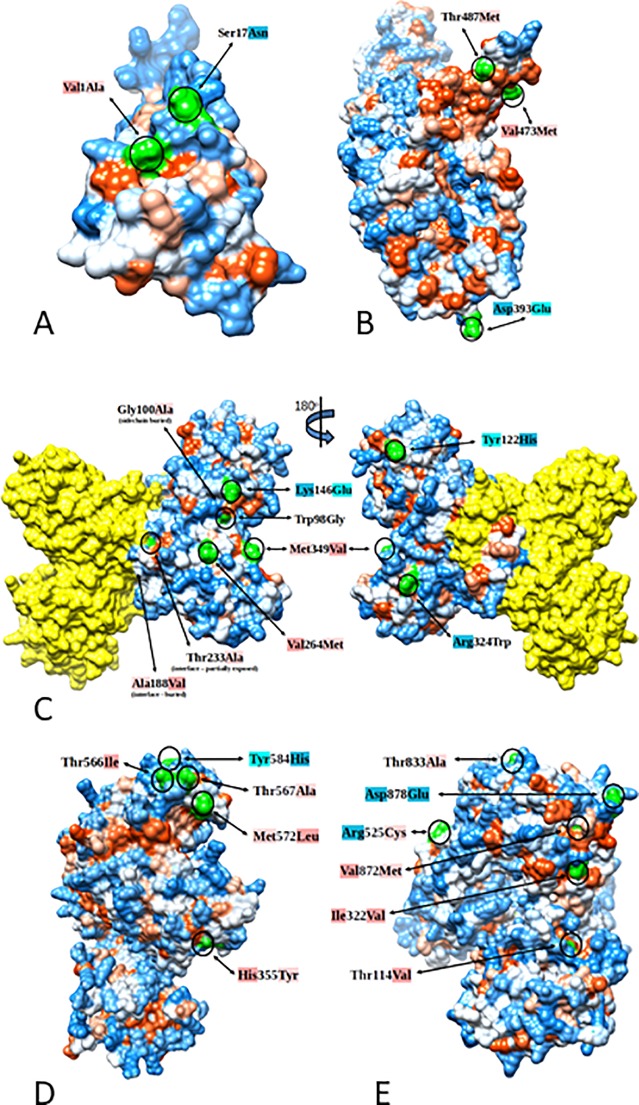
The question of how Zika virus (ZIKV) changed from a seemingly mild virus to a human pathogen capable of microcephaly and sexual transmission remains unanswered. The unexpected emergence of ZIKV's pathogenicity and capacity for sexual transmission may be due to genetic changes, and future changes in phenotype may continue to occur as the virus expands its geographic range. Alternatively, the sheer size of the 2015-16 epidemic may have brought attention to a pre-existing virulent ZIKV phenotype in a highly susceptible population. Thus, it is important to identify patterns of genetic change that may yield a better understanding of ZIKV emergence and evolution. However, because ZIKV has an RNA genome and a polymerase incapable of proofreading, it undergoes rapid mutation which makes it difficult to identify combinations of mutations associated with viral emergence. As next generation sequencing technology has allowed whole genome consensus and variant sequence data to be generated for numerous virus samples, the task of analyzing these genomes for patterns of mutation has become more complex. However, understanding which combinations of mutations spread widely and become established in new geographic regions versus those that disappear relatively quickly is essential for defining the trajectory of an ongoing epidemic. In this study, multiscale analysis of the wealth of genomic data generated over the course of the epidemic combined with in vivo laboratory data allowed trends in mutations and outbreak trajectory to be assessed. Mutations were detected throughout the genome via deep sequencing, and many variants appeared in multiple samples and in some cases become consensus. Similarly, amino acids that were previously consensus in pre-outbreak samples were detected as low frequency variants in epidemic strains. Protein structural models indicate that most of the mutations associated with the epidemic transmission occur on the exposed surface of viral proteins. At the macroscale level, consensus data was organized into large and interactive databases to allow the spread of individual mutations and combinations of mutations to be visualized and assessed for temporal and geographical patterns. Thus, the use of multiscale modeling for identifying mutations or combinations of mutations that impact epidemic transmission and phenotypic impact can aid the formation of hypotheses which can then be tested using reverse genetics.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Comparative analysis of protein evolution in the genome of pre-epidemic and epidemic Zika virus.Infect Genet Evol. 2017 Jul;51:74-85. doi: 10.1016/j.meegid.2017.03.012. Epub 2017 Mar 14. Infect Genet Evol. 2017. PMID: 28315476
-
Zika virus in the Americas: Early epidemiological and genetic findings.Science. 2016 Apr 15;352(6283):345-349. doi: 10.1126/science.aaf5036. Epub 2016 Mar 24. Science. 2016. PMID: 27013429 Free PMC article.
-
Zika virus evolution and spread in the Americas.Nature. 2017 Jun 15;546(7658):411-415. doi: 10.1038/nature22402. Epub 2017 May 24. Nature. 2017. PMID: 28538734 Free PMC article.
-
Zika virus: The transboundary pathogen from mosquito and updates.Microb Pathog. 2018 Jan;114:476-482. doi: 10.1016/j.micpath.2017.12.031. Epub 2017 Dec 11. Microb Pathog. 2018. PMID: 29241768 Review.
-
Zika virus-spread, epidemiology, genome, transmission cycle, clinical manifestation, associated challenges, vaccine and antiviral drug development.Virology. 2020 Apr;543:34-42. doi: 10.1016/j.virol.2020.01.015. Epub 2020 Feb 2. Virology. 2020. PMID: 32056845 Review.
Cited by
-
The clinical spectrum and immunopathological mechanisms underlying ZIKV-induced neurological manifestations.PLoS Negl Trop Dis. 2021 Aug 5;15(8):e0009575. doi: 10.1371/journal.pntd.0009575. eCollection 2021 Aug. PLoS Negl Trop Dis. 2021. PMID: 34351896 Free PMC article. Review.
-
Single Amino Acid Mutations Affect Zika Virus Replication In Vitro and Virulence In Vivo.Viruses. 2020 Nov 12;12(11):1295. doi: 10.3390/v12111295. Viruses. 2020. PMID: 33198111 Free PMC article.
-
Impact of Zika virus non-structural protein mutations on hippocampal damage.Neural Regen Res. 2025 Aug 1;20(8):2307-2308. doi: 10.4103/NRR.NRR-D-24-00493. Epub 2024 Jul 29. Neural Regen Res. 2025. PMID: 39359082 Free PMC article. No abstract available.
-
Hidden genomic diversity of SARS-CoV-2: implications for qRT-PCR diagnostics and transmission.bioRxiv [Preprint]. 2020 Jul 2:2020.07.02.184481. doi: 10.1101/2020.07.02.184481. bioRxiv. 2020. Update in: Genome Res. 2021 Apr;31(4):635-644. doi: 10.1101/gr.268961.120. PMID: 32637955 Free PMC article. Updated. Preprint.
-
A Small-Plaque Isolate of the Zika Virus with Envelope Domain III Mutations Affect Viral Entry and Replication in Mammalian but Not Mosquito Cells.Viruses. 2022 Feb 26;14(3):480. doi: 10.3390/v14030480. Viruses. 2022. PMID: 35336887 Free PMC article.
References
-
- Li C, Xu D, Ye Q, Hong S, Jiang Y, Liu X, et al. (2016) Zika Virus Disrupts Neural Progenitor Development and Leads to Microcephaly in Mice. Cell Stem Cell. - PubMed
-
- Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, et al. (2016) Zika Virus Associated with Microcephaly. New England Journal of Medicine 0: null. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
