

Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation
- PMID: 31813265
- PMCID: PMC7047114
- DOI: 10.1089/ars.2019.7963
Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation
Abstract
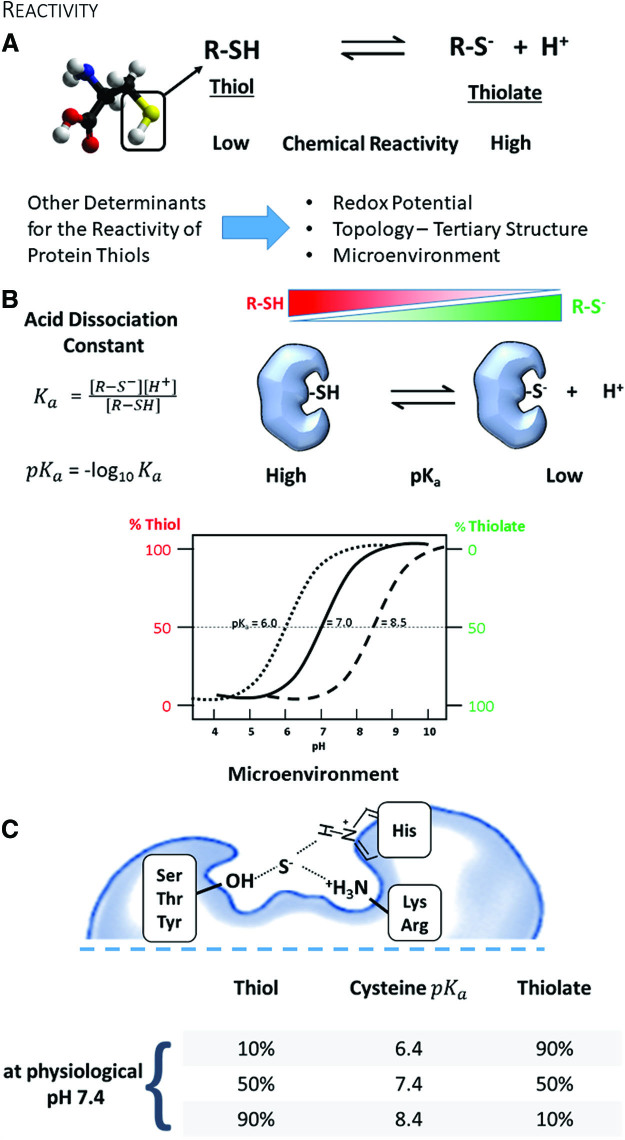
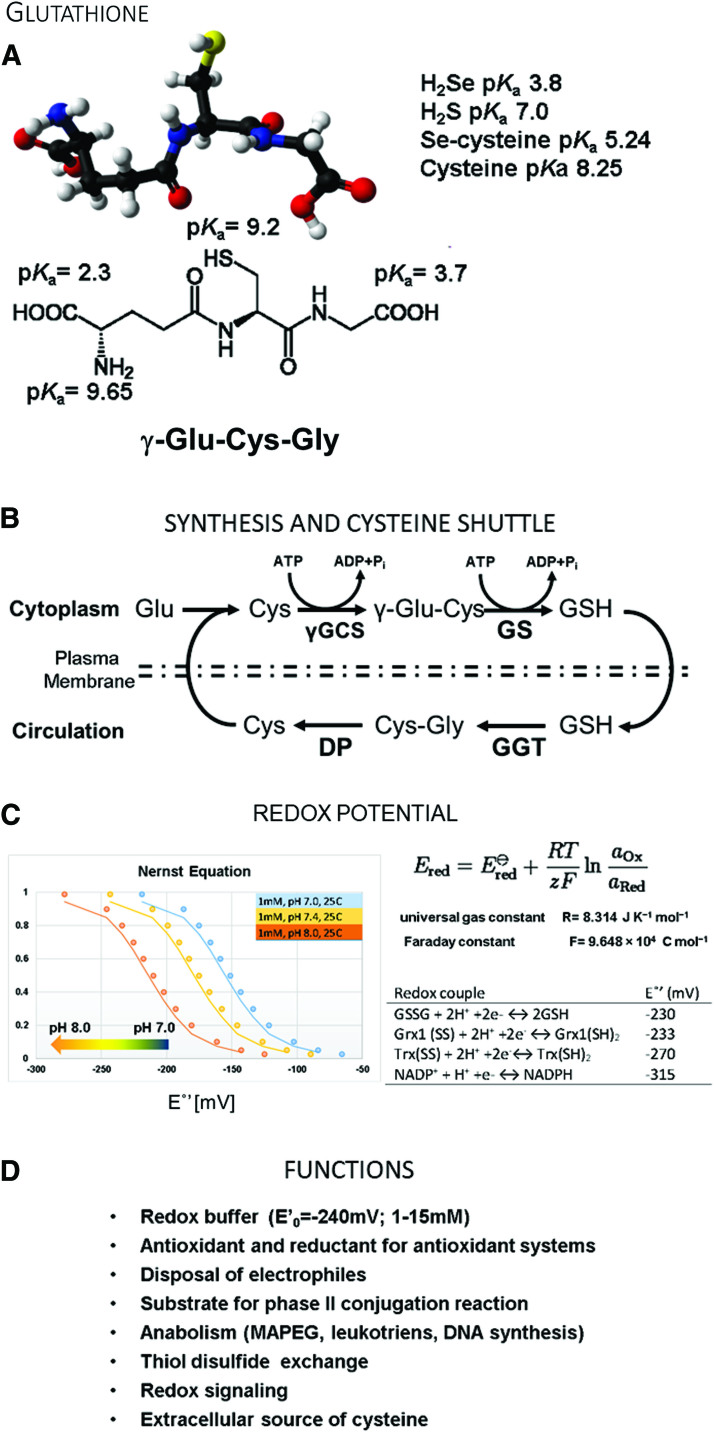
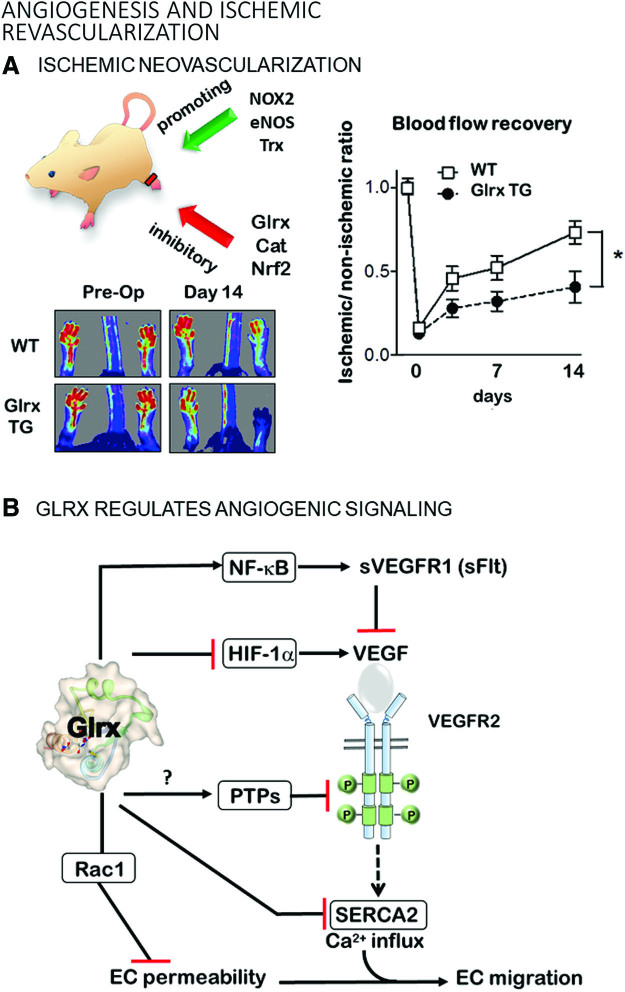
Significance: Over the past several years, oxidative post-translational modifications of protein cysteines have been recognized for their critical roles in physiology and pathophysiology. Cells have harnessed thiol modifications involving both oxidative and reductive steps for signaling and protein processing. One of these stages requires oxidation of cysteine to sulfenic acid, followed by two reduction reactions. First, glutathione (reduced glutathione [GSH]) forms a S-glutathionylated protein, and second, enzymatic or chemical reduction removes the modification. Under physiological conditions, these steps confer redox signaling and protect cysteines from irreversible oxidation. However, oxidative stress can overwhelm protein S-glutathionylation and irreversibly modify cysteine residues, disrupting redox signaling. Critical Issues: Glutaredoxins mainly catalyze the removal of protein-bound GSH and help maintain protein thiols in a highly reduced state without exerting direct antioxidant properties. Conversely, glutathione S-transferase (GST), peroxiredoxins, and occasionally glutaredoxins can also catalyze protein S-glutathionylation, thus promoting a dynamic redox environment. Recent Advances: The latest studies of glutaredoxin-1 (Glrx) transgenic or knockout mice demonstrate important distinct roles of Glrx in a variety of pathologies. Endogenous Glrx is essential to maintain normal hepatic lipid homeostasis and prevent fatty liver disease. Further, in vivo deletion of Glrx protects lungs from inflammation and bacterial pneumonia-induced damage, attenuates angiotensin II-induced cardiovascular hypertrophy, and improves ischemic limb vascularization. Meanwhile, exogenous Glrx administration can reverse pathological lung fibrosis. Future Directions: Although S-glutathionylation modifies many proteins, these studies suggest that S-glutathionylation and Glrx regulate specific pathways in vivo, and they implicate Glrx as a potential novel therapeutic target to treat diverse disease conditions. Antioxid. Redox Signal. 32, 677-700.
Keywords: NAFLD; NASH; fatty liver disease; hindlimb ischemia; peripheral artery disease.
Conflict of interest statement
The authors have nothing to disclose.
Figures





References
-
- Abdelsaid MA and El-Remessy AB. S-glutathionylation of LMW-PTP regulates VEGF-mediated FAK activation and endothelial cell migration. J Cell Sci 125: 4751–4760, 2012 - PubMed
-
- Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schöneich C, and Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 10: 1200–1207, 2004 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
