Current RNA-seq methodology reporting limits reproducibility
- PMID: 31813948
- PMCID: PMC7820846
- DOI: 10.1093/bib/bbz124
Current RNA-seq methodology reporting limits reproducibility
Abstract
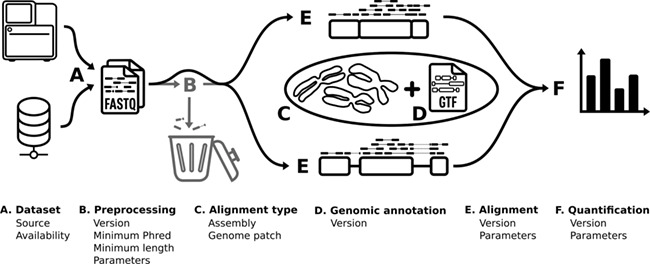
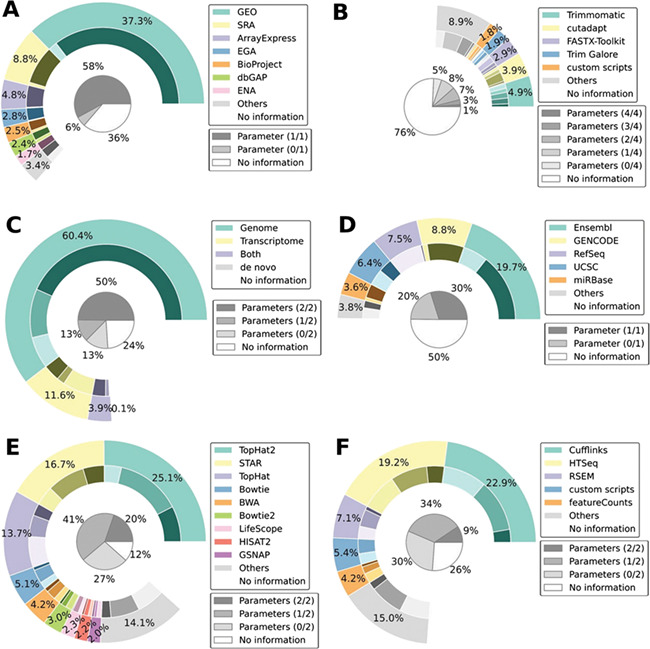
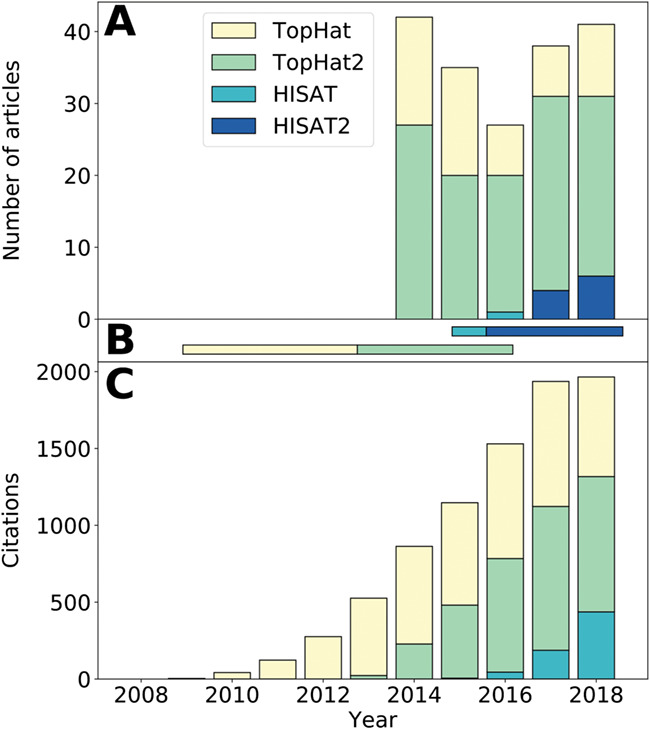
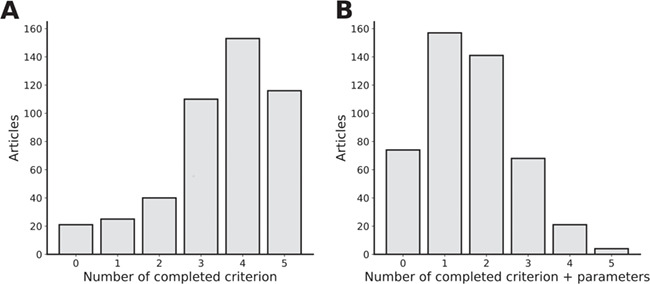
Ribonucleic acid sequencing (RNA-seq) identifies and quantifies RNA molecules from a biological sample. Transformation from raw sequencing data to meaningful gene or isoform counts requires an in silico bioinformatics pipeline. Such pipelines are modular in nature, built using selected software and biological references. Software is usually chosen and parameterized according to the sequencing protocol and biological question. However, while biological and technical noise is alleviated through replicates, biases due to the pipeline and choice of biological references are often overlooked. Here, we show that the current standard practice prevents reproducibility in RNA-seq studies by failing to specify required methodological information. Peer-reviewed articles are intended to apply currently accepted scientific and methodological standards. Inasmuch as the bias-less and optimal RNA-seq pipeline is not perfectly defined, methodological information holds a meaningful role in defining the results. This work illustrates the need for a standardized and explicit display of methodological information in RNA-seq experiments.
Keywords: RNA-sequencing; computational workflow; reproducibility.
© The Author(s) 2019. Published by Oxford University Press.
Figures