

Re-annotation of 191 developmental and epileptic encephalopathy-associated genes unmasks de novo variants in SCN1A
- PMID: 31814998
- PMCID: PMC6889285
- DOI: 10.1038/s41525-019-0106-7
Re-annotation of 191 developmental and epileptic encephalopathy-associated genes unmasks de novo variants in SCN1A
Abstract
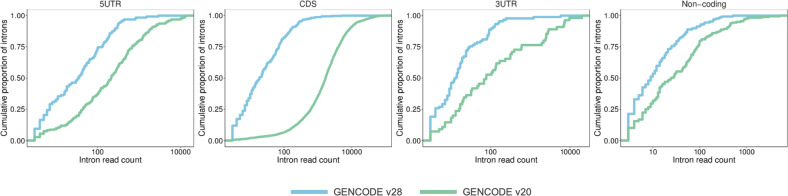
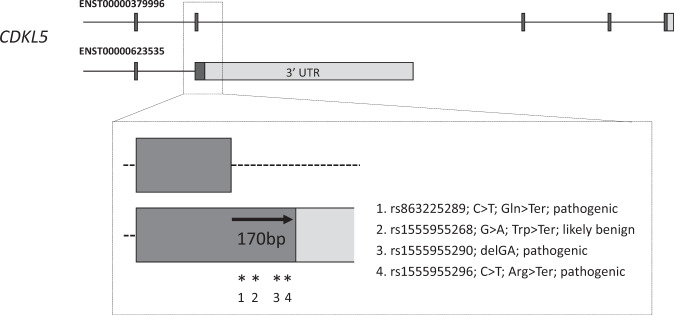
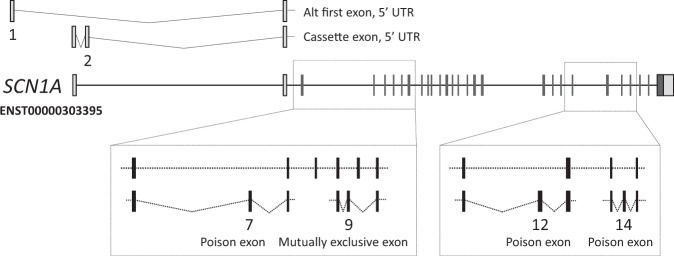
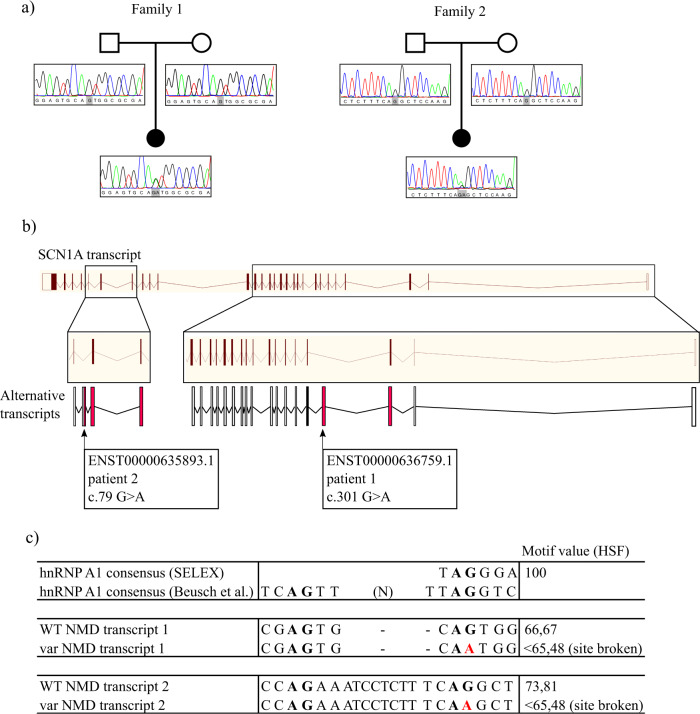
The developmental and epileptic encephalopathies (DEE) are a group of rare, severe neurodevelopmental disorders, where even the most thorough sequencing studies leave 60-65% of patients without a molecular diagnosis. Here, we explore the incompleteness of transcript models used for exome and genome analysis as one potential explanation for a lack of current diagnoses. Therefore, we have updated the GENCODE gene annotation for 191 epilepsy-associated genes, using human brain-derived transcriptomic libraries and other data to build 3,550 putative transcript models. Our annotations increase the transcriptional 'footprint' of these genes by over 674 kb. Using SCN1A as a case study, due to its close phenotype/genotype correlation with Dravet syndrome, we screened 122 people with Dravet syndrome or a similar phenotype with a panel of exon sequences representing eight established genes and identified two de novo SCN1A variants that now - through improved gene annotation - are ascribed to residing among our exons. These two (from 122 screened people, 1.6%) molecular diagnoses carry significant clinical implications. Furthermore, we identified a previously classified SCN1A intronic Dravet syndrome-associated variant that now lies within a deeply conserved exon. Our findings illustrate the potential gains of thorough gene annotation in improving diagnostic yields for genetic disorders.
Keywords: Medical genomics; Molecular medicine.
© The Author(s) 2019.
Conflict of interest statement
Competing interestsC.A.S., A.S.R. and N.J.L. are employed by Congenica Ltd. P.F. is a member of the scientific advisory boards for Fabric Genomics, Inc., and Eagle Genomics, Ltd. S.P. is an employee and D.V. is a postdoc of AstraZeneca. The remaining authors declare they have no competing interests.
Figures
References
-
- Mark, C. et al. The 100,000 Genomes Project Protocol (2017).
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
