

Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy
- PMID: 31815659
- PMCID: PMC6902404
- DOI: 10.1186/s13045-019-0818-2
Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy
Abstract
The biggest hurdle to targeted cancer therapy is the inevitable emergence of drug resistance. Tumor cells employ different mechanisms to resist the targeting agent. Most commonly in EGFR-mutant non-small cell lung cancer, secondary resistance mutations on the target kinase domain emerge to diminish the binding affinity of first- and second-generation inhibitors. Other alternative resistance mechanisms include activating complementary bypass pathways and phenotypic transformation. Sequential monotherapies promise to temporarily address the problem of acquired drug resistance, but evidently are limited by the tumor cells' ability to adapt and evolve new resistance mechanisms to persist in the drug environment. Recent studies have nominated a model of drug resistance and tumor progression under targeted therapy as a result of a small subpopulation of cells being able to endure the drug (minimal residual disease cells) and eventually develop further mutations that allow them to regrow and become the dominant population in the therapy-resistant tumor. This subpopulation of cells appears to have developed through a subclonal event, resulting in driver mutations different from the driver mutation that is tumor-initiating in the most common ancestor. As such, an understanding of intratumoral heterogeneity-the driving force behind minimal residual disease-is vital for the identification of resistance drivers that results from branching evolution. Currently available methods allow for a more comprehensive and holistic analysis of tumor heterogeneity in that issues associated with spatial and temporal heterogeneity can now be properly addressed. This review provides some background regarding intratumoral heterogeneity and how it leads to incomplete molecular response to targeted therapies, and proposes the use of single-cell methods, sequential liquid biopsy, and multiregion sequencing to discover the link between intratumoral heterogeneity and early adaptive drug resistance. In summary, minimal residual disease as a result of intratumoral heterogeneity is the earliest form of acquired drug resistance. Emerging technologies such as liquid biopsy and single-cell methods allow for studying targetable drivers of minimal residual disease and contribute to preemptive combinatorial targeting of both drivers of the tumor and its minimal residual disease cells.
Keywords: Acquired drug resistance; Adaptive evolution; Minimal residual disease; Tumor heterogeneity.
Conflict of interest statement
The authors declare the following conflict of interest: Speaker Bureau for Merck, AstraZeneca and Bayer, and Advisory Board consultant for AstraZeneca and Apollomics (PCM).
Figures
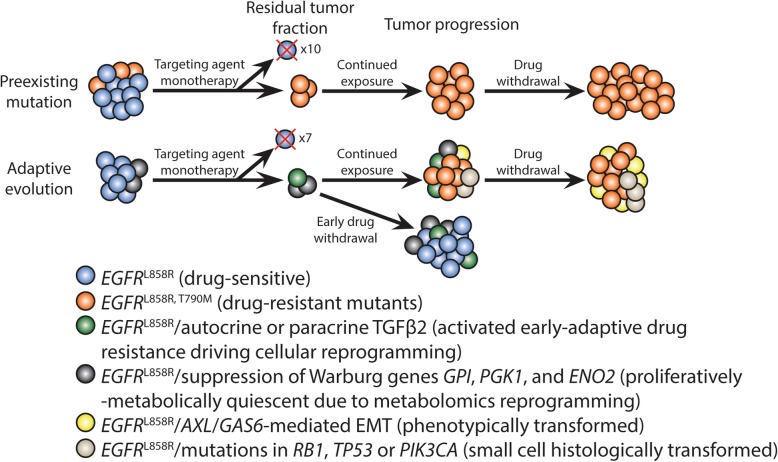
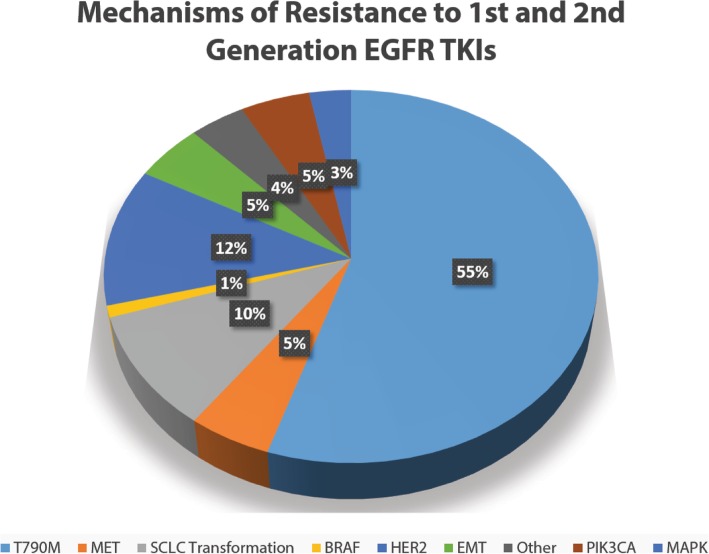
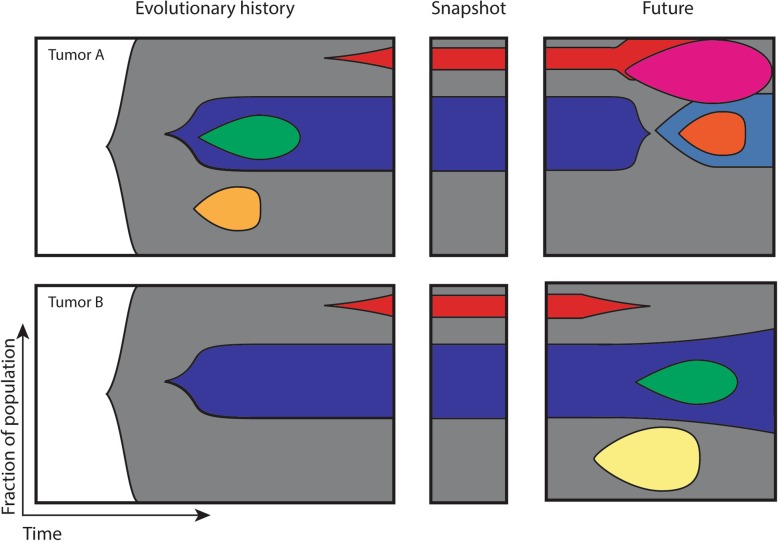
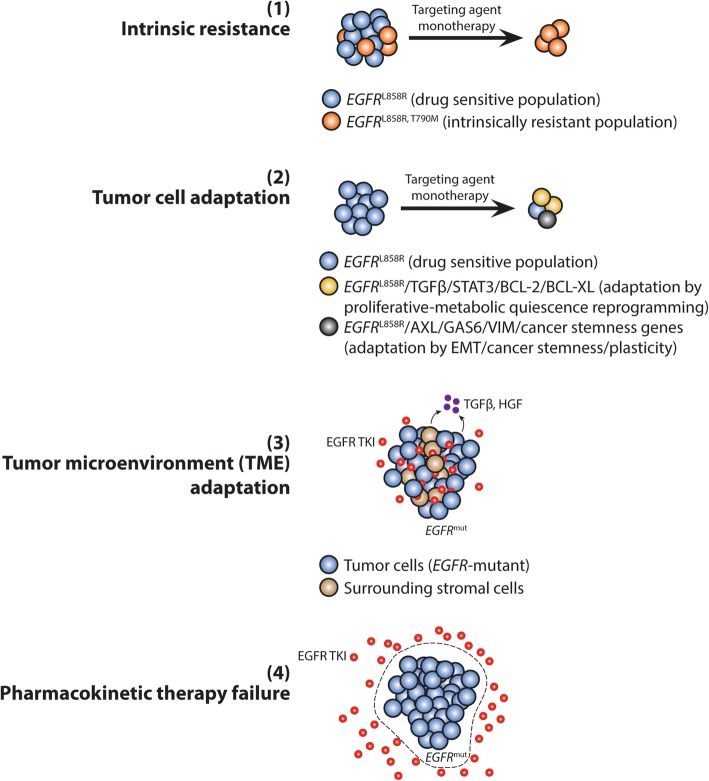
References
-
- Ueda K, Suekane S, Nishihara K, Ogasawara N, Kurose H, Hayashi S, et al. Duration of first-line treatment with molecular targeted-therapy is a prognostic factor in metastatic renal cell carcinoma. Anticancer Res. 2015;35(6):3415–3421. - PubMed
-
- Casadio C, Guarize J, Donghi S, Di Tonno C, Fumagalli C, Vacirca D, et al. Molecular testing for targeted therapy in advanced non-small cell lung cancer: suitability of endobronchial ultrasound transbronchial needle aspiration. Am J Clin Pathol. 2015;144(4):629–634. doi: 10.1309/AJCPXGRAIMB4CTQ3. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
