

Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis
- PMID: 31817302
- PMCID: PMC6947644
- DOI: 10.3390/genes10121005
Transcriptome Meta-Analysis Deciphers a Dysregulation in Immune Response-Associated Gene Signatures during Sepsis
Abstract
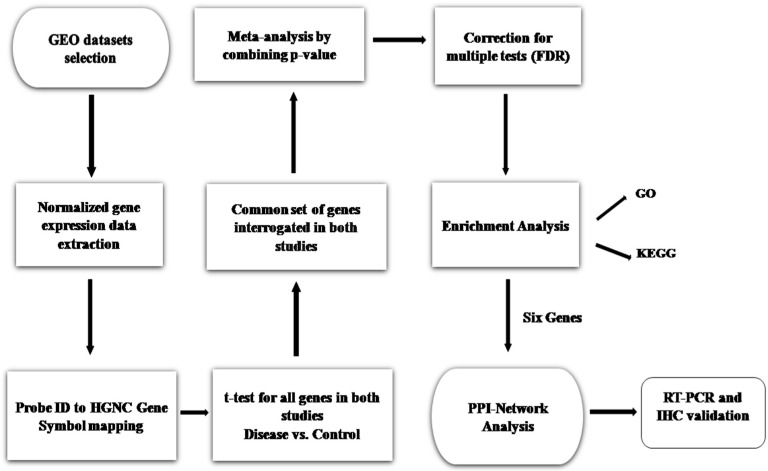
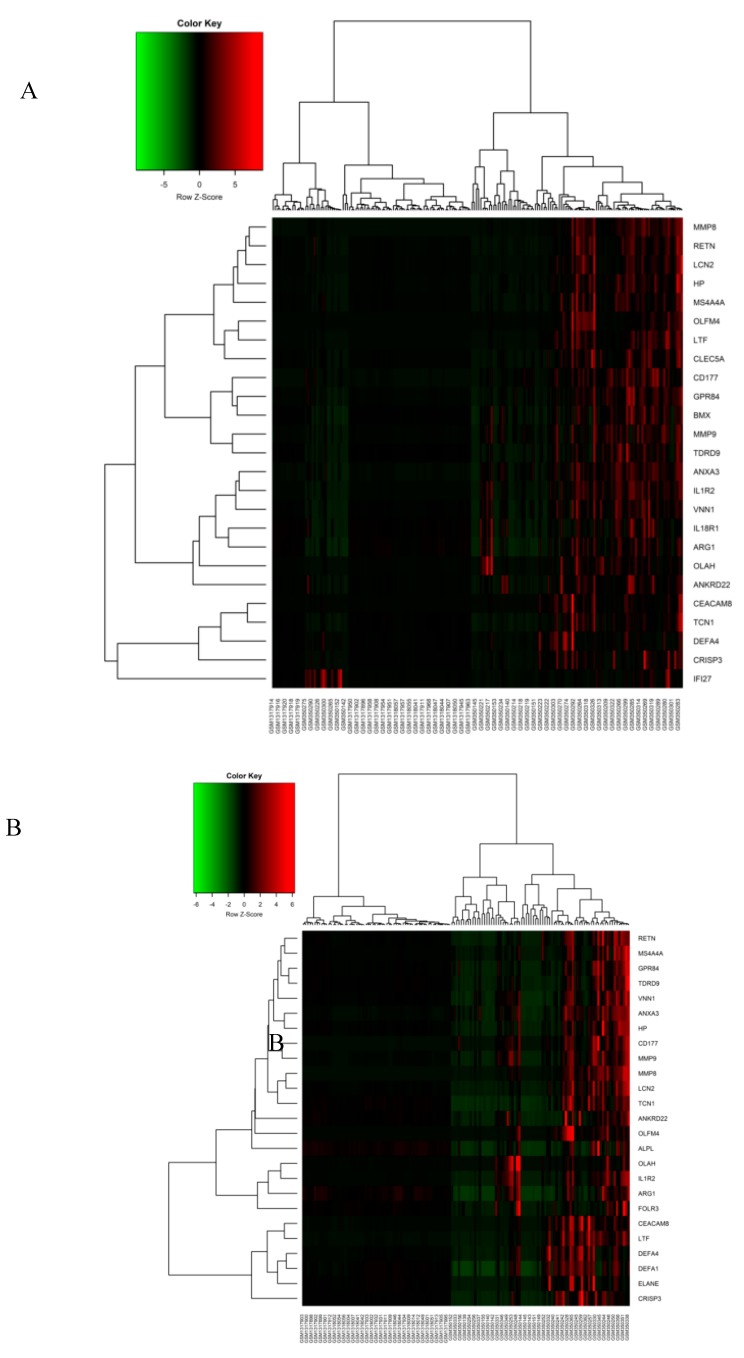
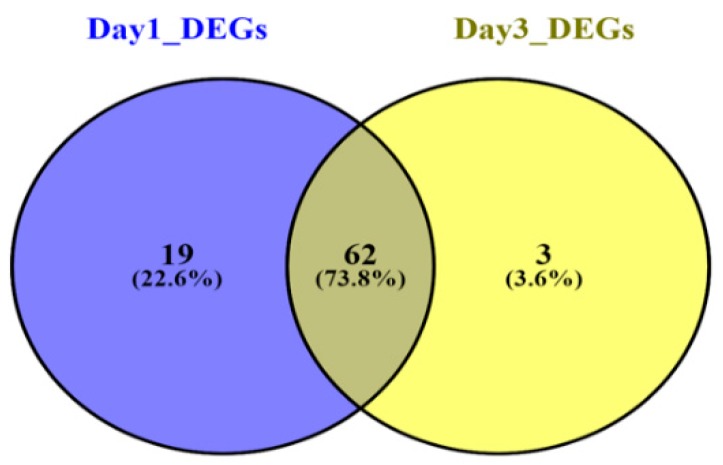
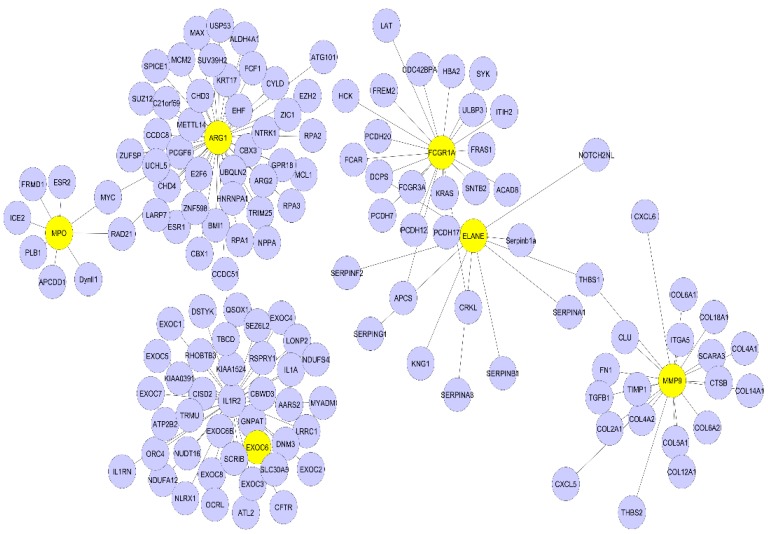
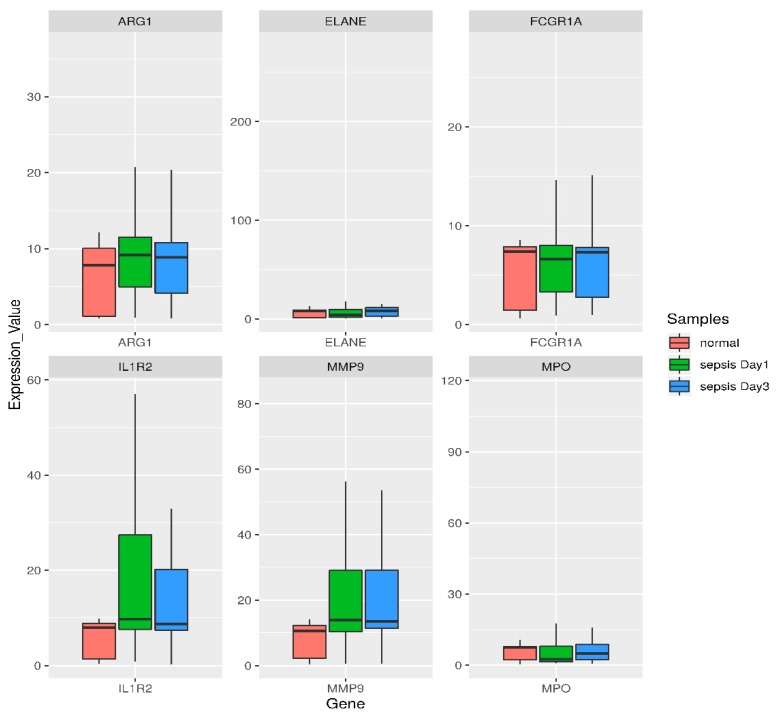
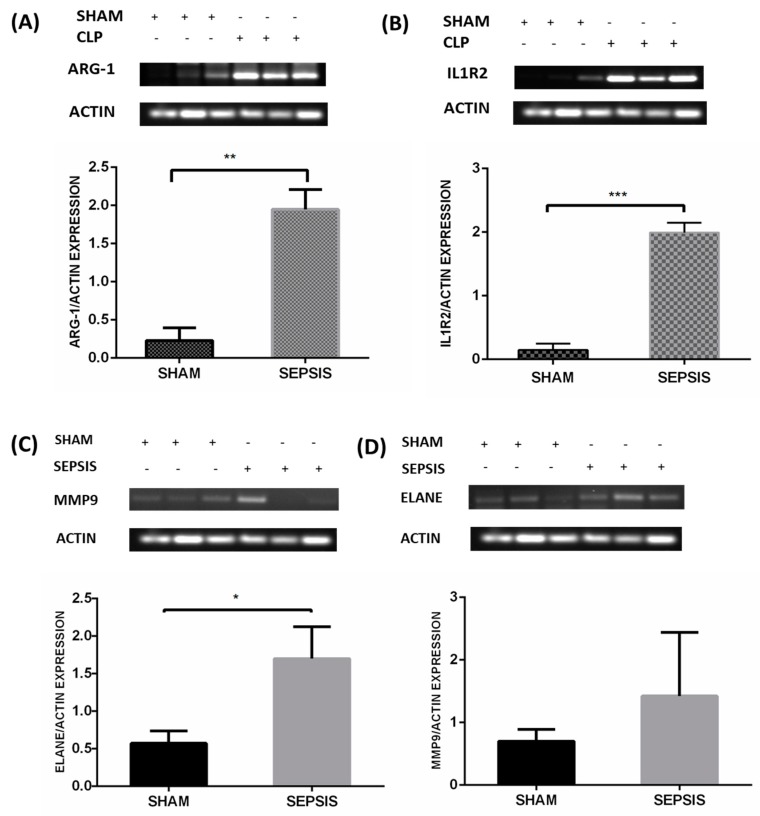
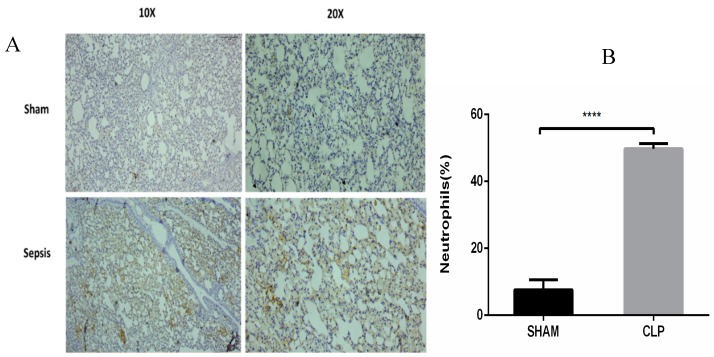
Sepsis is a life-threatening disease induced by a systemic inflammatory response, which leads to organ dysfunction and mortality. In sepsis, the host immune response is depressed and unable to cope with infection; no drug is currently available to treat this. The lungs are frequently the starting point for sepsis. This study aimed to identify potential genes for diagnostics and therapeutic purposes in sepsis by a comprehensive bioinformatics analysis. Our criteria are to unravel sepsis-associated signature genes from gene expression datasets. Differentially expressed genes (DEGs) were identified from samples of sepsis patients using a meta-analysis and then further subjected to functional enrichment and protein‒protein interaction (PPI) network analysis for examining their potential functions. Finally, the expression of the topmost upregulated genes (ARG1, IL1R2, ELANE, MMP9) was quantified by reverse transcriptase-PCR (RT-PCR), and myeloperoxidase (MPO) expression was confirmed by immunohistochemistry (IHC) staining in the lungs of a well-established sepsis mouse model. We found that all the four genes were upregulated in semiquantitative RT-PCR studies; however, MMP9 showed a nonsignificant increase in expression. MPO staining showed strong immunoreactivity in sepsis as compared to the control. This study demonstrates the role of significant and widespread immune activation (IL1R2, MMP9), along with oxidative stress (ARG1) and the recruitment of neutrophils, in sepsis (ELANE, MPO).
Keywords: DEG; PPI; meta-analysis; sepsis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Singer M., Deutschman C.S., Seymour C.W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G.R., Chiche J.D., Coopersmith C.M., et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. - DOI - PMC - PubMed
-
- Fleischmann C., Scherag A., Adhikari N.K., Hartog C.S., Tsaganos T., Schlattmann P., Angus D.C., Reinhart K. Assessment of Global Incidence and Mortality of Hospital-treated Sepsis. Current Estimates and Limitations. Am. J. Respir. Crit. Care Med. 2016;193:259–272. doi: 10.1164/rccm.201504-0781OC. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
