

GLUL Ablation Can Confer Drug Resistance to Cancer Cells via a Malate-Aspartate Shuttle-Mediated Mechanism
- PMID: 31817360
- PMCID: PMC6966511
- DOI: 10.3390/cancers11121945
GLUL Ablation Can Confer Drug Resistance to Cancer Cells via a Malate-Aspartate Shuttle-Mediated Mechanism
Abstract
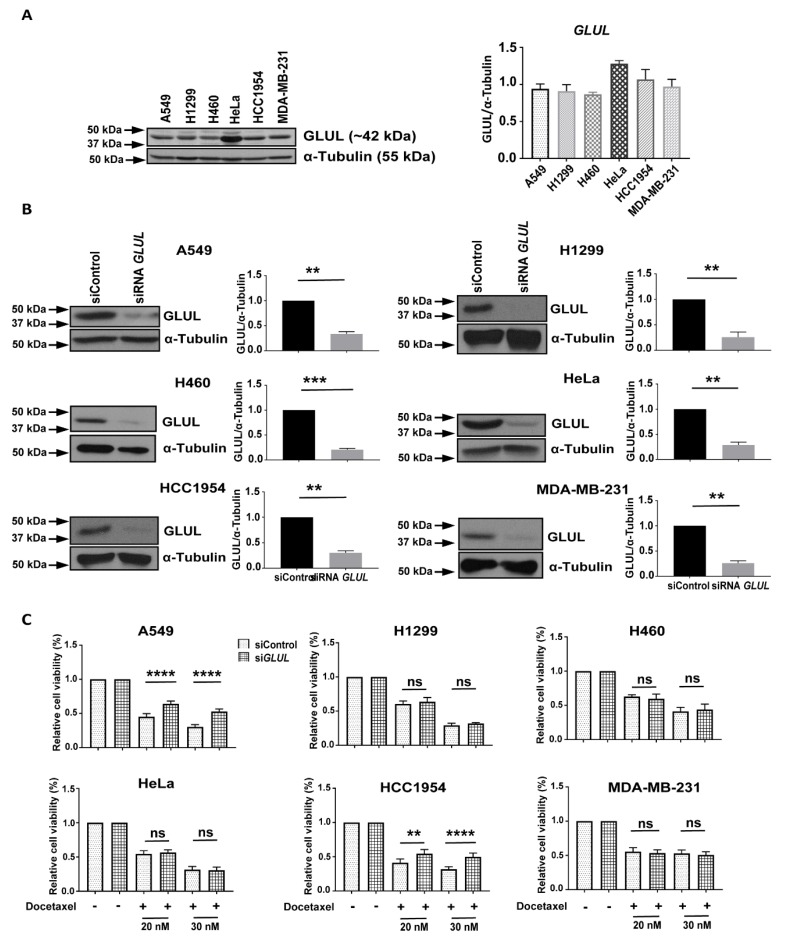
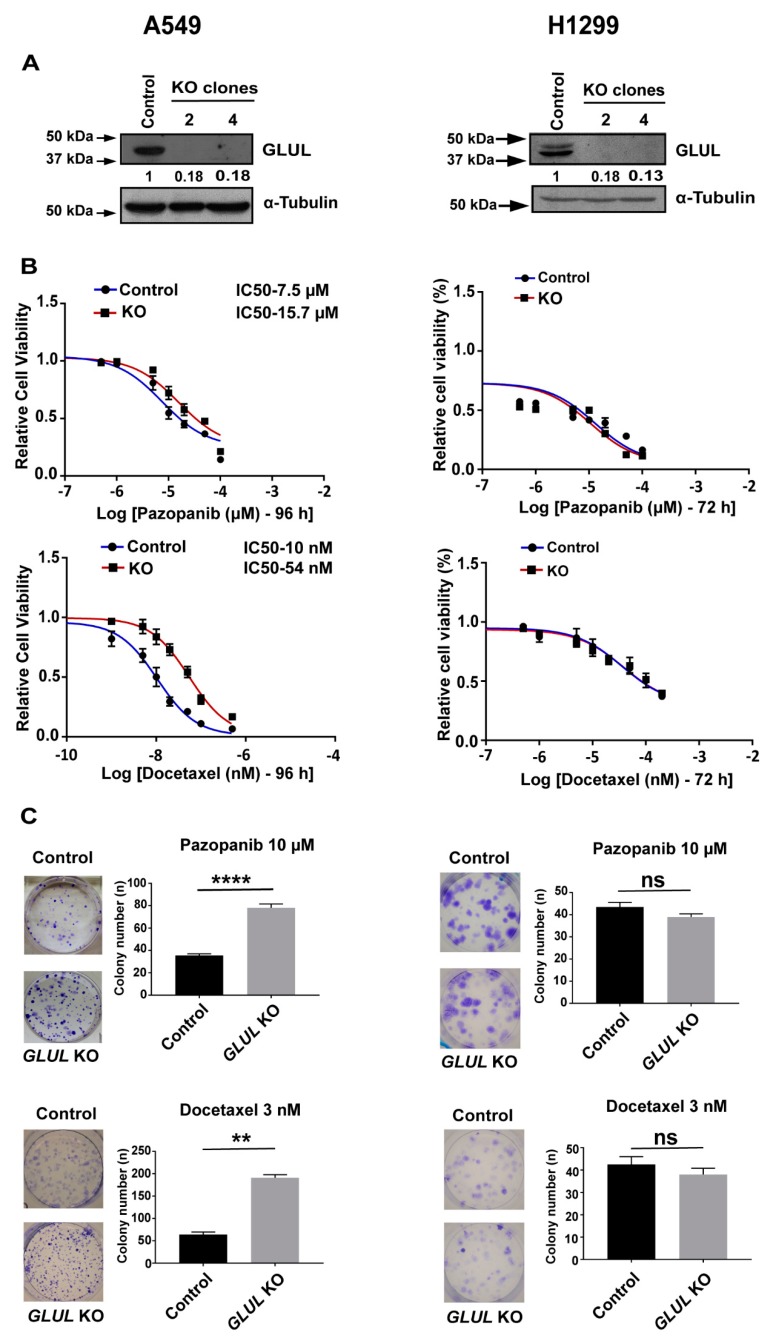
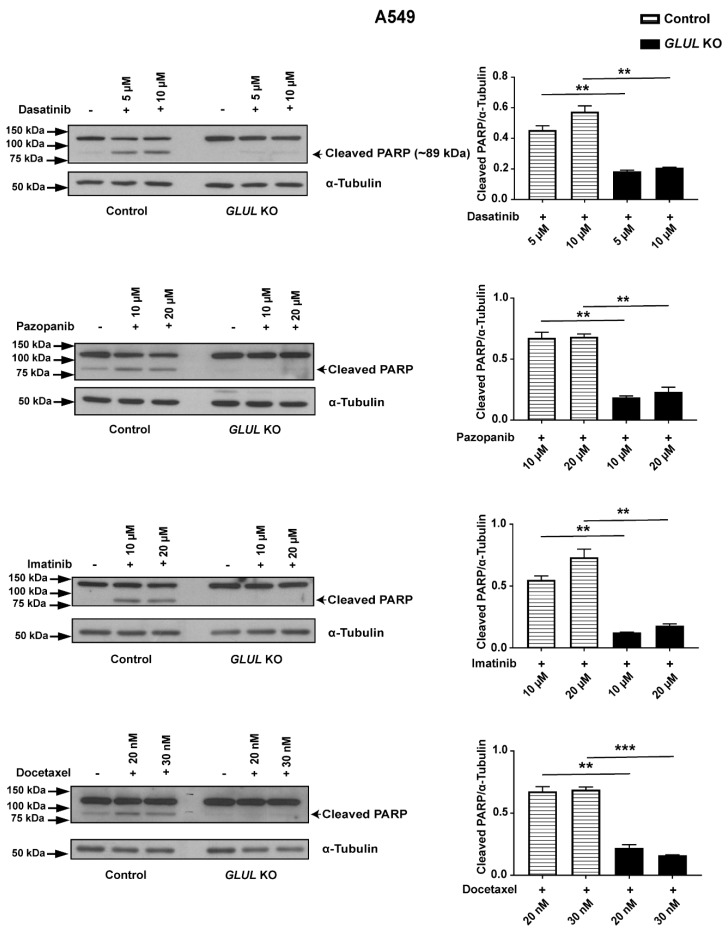
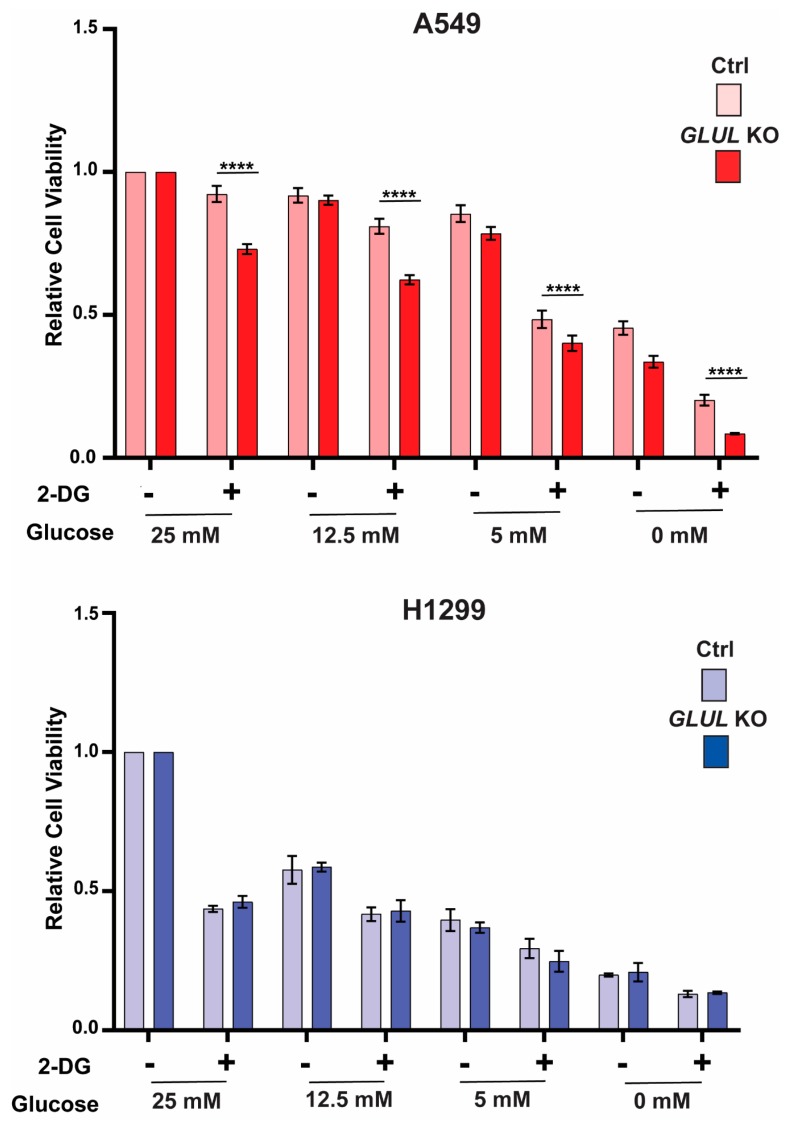
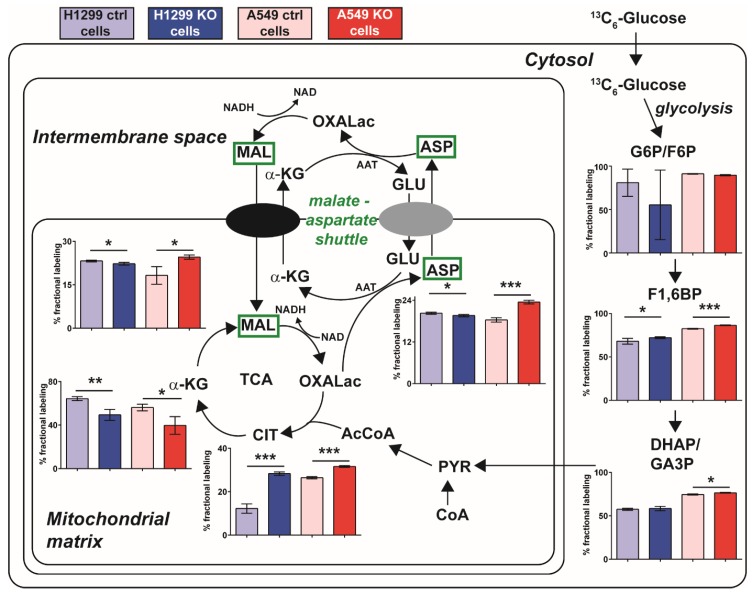
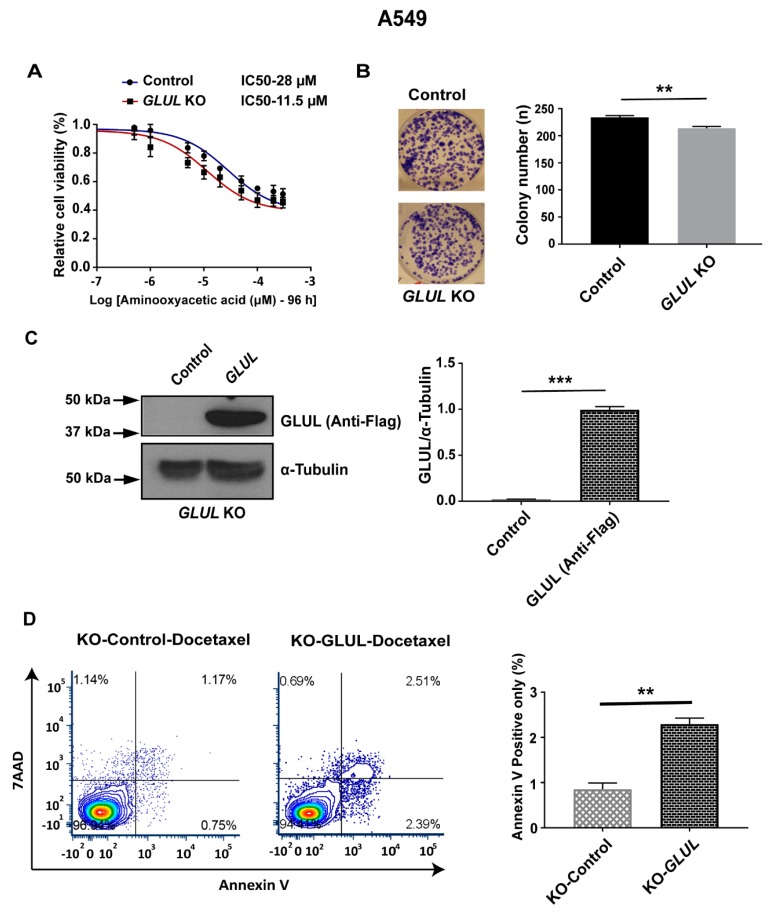
Glutamate-ammonia ligase (GLUL) is important for acid-base homeostasis, ammonia detoxification, cell signaling, and proliferation. Here, we reported that GLUL ablation conferred resistance to several anticancer drugs in specific cancer cell lines while leaving other cell lines non-resistant to the same drugs. To understand the biochemical mechanics supporting this drug resistance, we compared drug-resistant GLUL knockout (KO) A549 non-small-cell lung carcinoma (NSCLC) cells with non-resistant GLUL KO H1299 NSCLC cells and found that the resistant A549 cells, to a larger extent, depended on exogenous glucose for proliferation. As GLUL activity is linked to the tricarboxylic acid (TCA) cycle via reversed glutaminolysis, we probed carbon flux through both glycolysis and TCA pathways by means of 13C5 glutamine, 13C5 glutamate, and 13C6 glucose tracing. We observed increased labeling of malate and aspartate in A549 GLUL KO cells, whereas the non-resistant GLUL KO H1299 cells displayed decreased 13C-labeling. The malate and aspartate shuttle supported cellular NADH production and was associated with cellular metabolic fitness. Inhibition of the malate-aspartate shuttle with aminooxyacetic acid significantly impacted upon cell viability with an IC50 of 11.5 μM in resistant GLUL KO A549 cells compared to 28 μM in control A549 cells, linking resistance to the malate-aspartate shuttle. Additionally, rescuing GLUL expression in A549 KO cells increased drug sensitivity. We proposed a novel metabolic mechanism in cancer drug resistance where the increased capacity of the malate-aspartate shuttle increased metabolic fitness, thereby facilitating cancer cells to escape drug pressure.
Keywords: GLUL; LC-MS; NSCLC; drug resistance; glutamine; glycolysis; metabolism; metabolomics; targeted metabolomics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Increased glutamine anabolism sensitizes non-small cell lung cancer to gefitinib treatment.Cell Death Discov. 2018 Aug 9;4:24. doi: 10.1038/s41420-018-0086-x. eCollection 2018. Cell Death Discov. 2018. PMID: 30109143 Free PMC article.
-
Probing the cardiac malate-aspartate shuttle non-invasively using hyperpolarized [1,2-13 C2 ]pyruvate.NMR Biomed. 2018 Jan;31(1). doi: 10.1002/nbm.3845. Epub 2017 Nov 6. NMR Biomed. 2018. PMID: 29106770
-
The cardioprotective effect of sildenafil is mediated by the activation of malate dehydrogenase and an increase in the malate-aspartate shuttle in cardiomyocytes.Biochem Pharmacol. 2017 Mar 1;127:60-70. doi: 10.1016/j.bcp.2016.12.017. Epub 2016 Dec 23. Biochem Pharmacol. 2017. PMID: 28017777
-
Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: current evidence and pharmacological tools.Biochem Pharmacol. 2006 Feb 14;71(4):399-407. doi: 10.1016/j.bcp.2005.10.011. Epub 2005 Dec 20. Biochem Pharmacol. 2006. PMID: 16368075 Review.
-
TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma.Cancers (Basel). 2019 Dec 25;12(1):68. doi: 10.3390/cancers12010068. Cancers (Basel). 2019. PMID: 31881713 Free PMC article. Review.
Cited by
-
Effect of Short-Chain Fatty Acids and Polyunsaturated Fatty Acids on Metabolites in H460 Lung Cancer Cells.Molecules. 2023 Mar 3;28(5):2357. doi: 10.3390/molecules28052357. Molecules. 2023. PMID: 36903601 Free PMC article.
-
GLUL stabilizes N-Cadherin by antagonizing β-Catenin to inhibit the progresses of gastric cancer.Acta Pharm Sin B. 2024 Feb;14(2):698-711. doi: 10.1016/j.apsb.2023.11.008. Epub 2023 Nov 9. Acta Pharm Sin B. 2024. PMID: 38322340 Free PMC article.
-
Glutamine Synthetase as a Therapeutic Target for Cancer Treatment.Int J Mol Sci. 2021 Feb 8;22(4):1701. doi: 10.3390/ijms22041701. Int J Mol Sci. 2021. PMID: 33567690 Free PMC article. Review.
-
N-phenylmaleimide induces bioenergetic switch and suppresses tumor growth in glioblastoma tumorspheres by inhibiting SLC25A11.Cancer Cell Int. 2025 May 22;25(1):184. doi: 10.1186/s12935-025-03813-y. Cancer Cell Int. 2025. PMID: 40405188 Free PMC article.
-
Glutamine synthetase regulates the immune microenvironment and cancer development through the inflammatory pathway.Int J Med Sci. 2023 Jan 1;20(1):35-49. doi: 10.7150/ijms.75625. eCollection 2023. Int J Med Sci. 2023. PMID: 36619229 Free PMC article.
References
-
- Patel C., Stenke L., Varma S., Lindberg M.L., Bjorkholm M., Sjoberg J., Viktorsson K., Lewensohn R., Landgren O., Gottesman M.M., et al. Multidrug resistance in relapsed acute myeloid leukemia: EVIDENCE of biological heterogeneity. Cancer. 2013;119:3076–3083. doi: 10.1002/cncr.28098. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
