

Single-Cell RNA Sequencing Reveals Renal Endothelium Heterogeneity and Metabolic Adaptation to Water Deprivation
- PMID: 31818909
- PMCID: PMC6935008
- DOI: 10.1681/ASN.2019080832
Single-Cell RNA Sequencing Reveals Renal Endothelium Heterogeneity and Metabolic Adaptation to Water Deprivation
Abstract
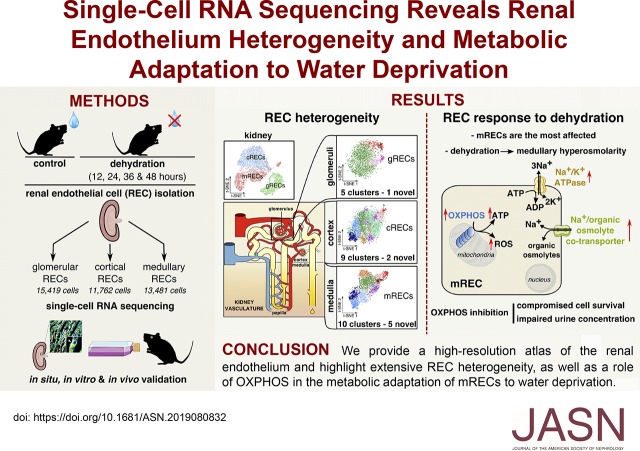
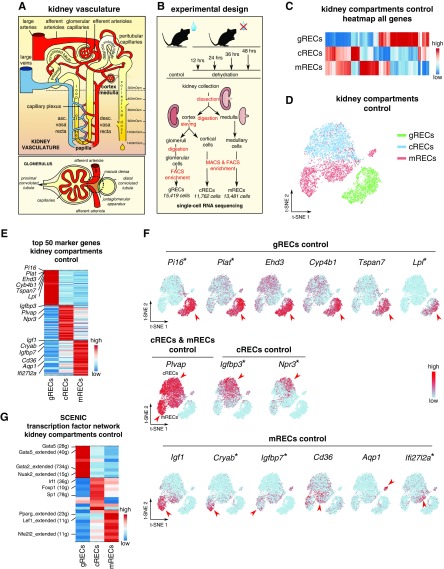
Background: Renal endothelial cells from glomerular, cortical, and medullary kidney compartments are exposed to different microenvironmental conditions and support specific kidney processes. However, the heterogeneous phenotypes of these cells remain incompletely inventoried. Osmotic homeostasis is vitally important for regulating cell volume and function, and in mammals, osmotic equilibrium is regulated through the countercurrent system in the renal medulla, where water exchange through endothelium occurs against an osmotic pressure gradient. Dehydration exposes medullary renal endothelial cells to extreme hyperosmolarity, and how these cells adapt to and survive in this hypertonic milieu is unknown.
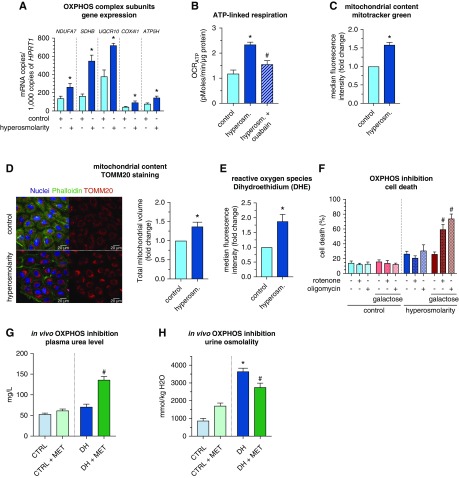
Methods: We inventoried renal endothelial cell heterogeneity by single-cell RNA sequencing >40,000 mouse renal endothelial cells, and studied transcriptome changes during osmotic adaptation upon water deprivation. We validated our findings by immunostaining and functionally by targeting oxidative phosphorylation in a hyperosmolarity model in vitro and in dehydrated mice in vivo.
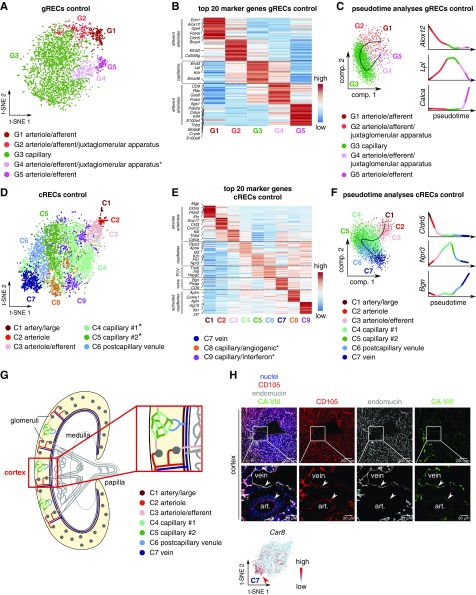
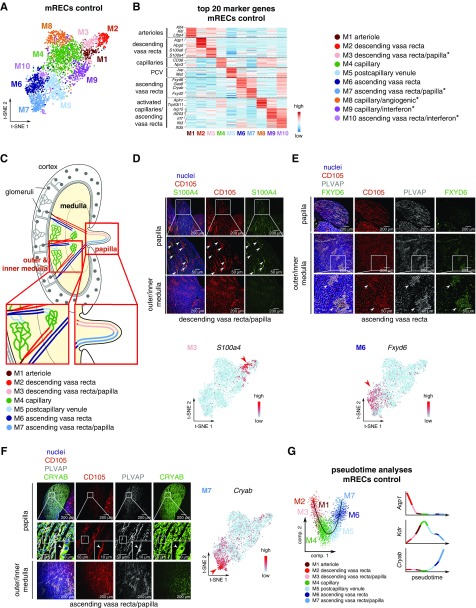
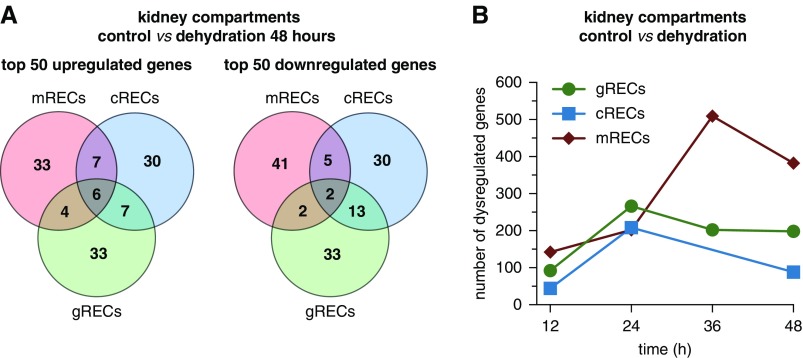
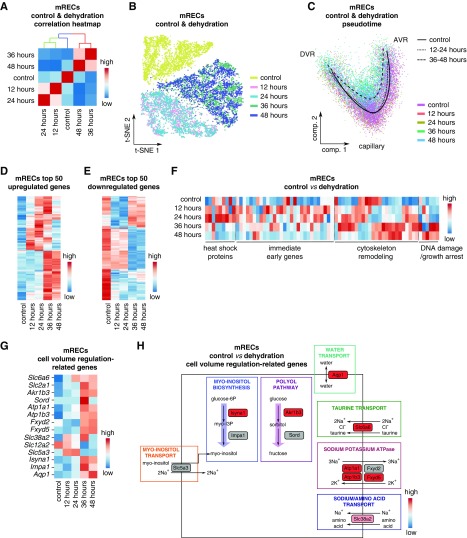
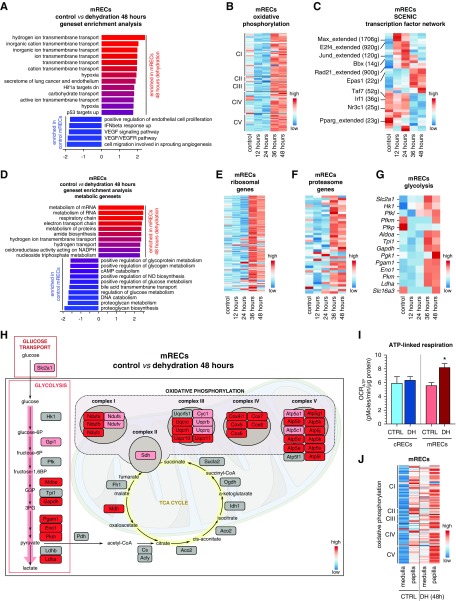
Results: We identified 24 renal endothelial cell phenotypes (of which eight were novel), highlighting extensive heterogeneity of these cells between and within the cortex, glomeruli, and medulla. In response to dehydration and hypertonicity, medullary renal endothelial cells upregulated the expression of genes involved in the hypoxia response, glycolysis, and-surprisingly-oxidative phosphorylation. Endothelial cells increased oxygen consumption when exposed to hyperosmolarity, whereas blocking oxidative phosphorylation compromised endothelial cell viability during hyperosmotic stress and impaired urine concentration during dehydration.
Conclusions: This study provides a high-resolution atlas of the renal endothelium and highlights extensive renal endothelial cell phenotypic heterogeneity, as well as a previously unrecognized role of oxidative phosphorylation in the metabolic adaptation of medullary renal endothelial cells to water deprivation.
Keywords: dehydration; heterogeneity; oxidative phosphorylation; renal endothelial cells; scRNA-sequencing; urine concentration.
Copyright © 2020 by the American Society of Nephrology.
Figures
Comment in
-
Endothelial Cell Identity, Heterogeneity and Plasticity in the Kidney.J Am Soc Nephrol. 2020 Jan;31(1):1-2. doi: 10.1681/ASN.2019111179. Epub 2019 Dec 9. J Am Soc Nephrol. 2020. PMID: 31821991 Free PMC article. No abstract available.
-
Endothelium adapts to survive dehydration.Nat Rev Nephrol. 2020 Apr;16(4):187. doi: 10.1038/s41581-020-0250-4. Nat Rev Nephrol. 2020. PMID: 31959940 No abstract available.
References
-
- Molema G, Aird WC: Vascular heterogeneity in the kidney. Semin Nephrol 32: 145–155, 2012 - PubMed
-
- Jourde-Chiche N, Fakhouri F, Dou L, Bellien J, Burtey S, Frimat M, et al. .: Endothelium structure and function in kidney health and disease. Nat Rev Nephrol 15: 87–108, 2019 - PubMed
-
- Kriz W: Fenestrated glomerular capillaries are unique. J Am Soc Nephrol 19: 1439–1440, 2008 - PubMed
-
- Eckardt KU, Bernhardt WM, Weidemann A, Warnecke C, Rosenberger C, Wiesener MS, et al. .: Role of hypoxia in the pathogenesis of renal disease. Kidney Int Suppl S46–S51, 2005 - PubMed
-
- Pallone TL, Edwards A, Mattson DL: Renal medullary circulation. Compr Physiol 2: 97–140, 2012 - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
