

Antibody validation for Western blot: By the user, for the user
- PMID: 31819006
- PMCID: PMC6983856
- DOI: 10.1074/jbc.RA119.010472
Antibody validation for Western blot: By the user, for the user
Abstract
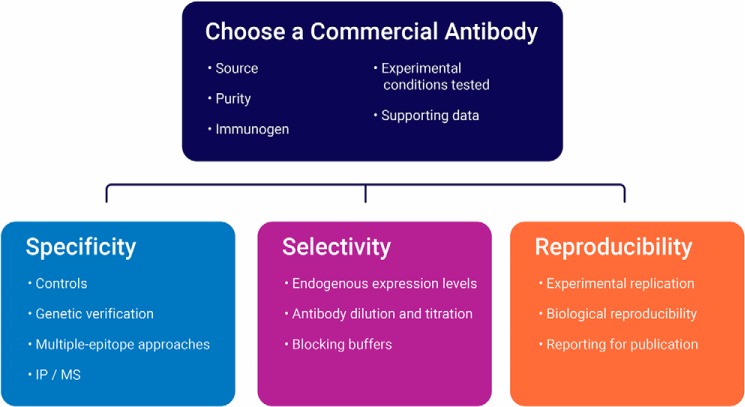
Well-characterized antibody reagents play a key role in the reproducibility of research findings, and inconsistent antibody performance leads to variability in Western blotting and other immunoassays. The current lack of clear, accepted standards for antibody validation and reporting of experimental details contributes to this problem. Because the performance of primary antibodies is strongly influenced by assay context, recommendations for validation and usage are unique to each type of immunoassay. Practical strategies are proposed for the validation of primary antibody specificity, selectivity, and reproducibility using Western blot analysis. The antibody should produce reproducible results within and between Western blotting experiments and the observed effect confirmed with a complementary or orthogonal method. Routine implementation of standardized antibody validation and reporting in immunoassays such as Western blotting may promote improved reproducibility across the global life sciences community.
Keywords: Immunoblot; Methodology; antibody; antibody validation; antigen; immunochemistry; irreproducibility crisis; protein domain; reproducibility; western blot.
© 2020 Pillai-Kastoori et al.
Conflict of interest statement
L. P.-K., S. D. S., and A. S.-G. are employees of LI-COR Biosciences, and S. H., A. R., and A. S. are employees of Abcam Plc
Figures
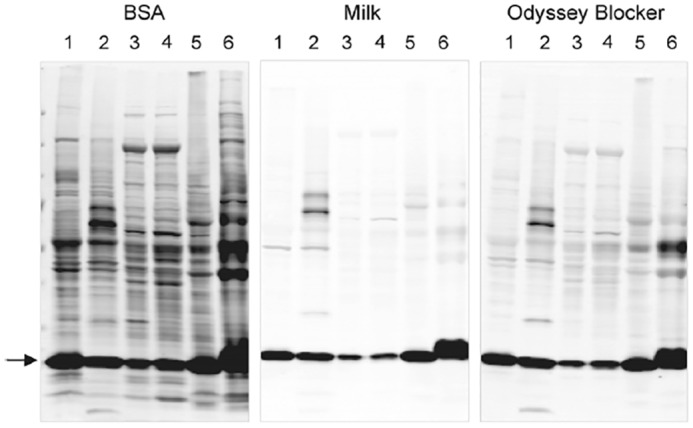
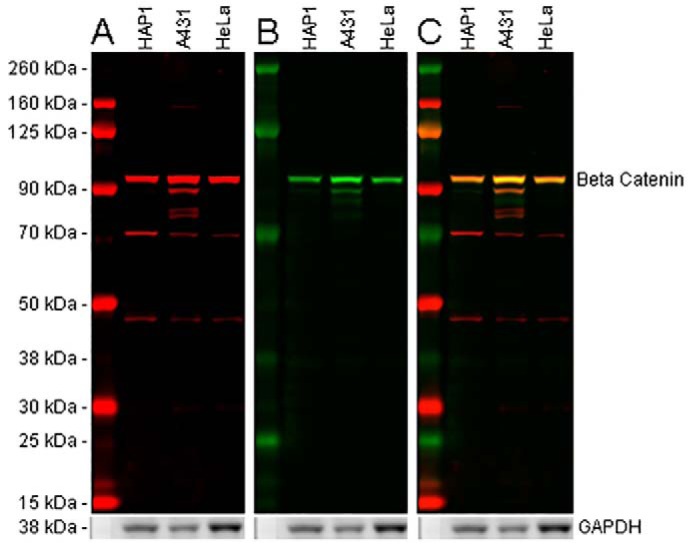
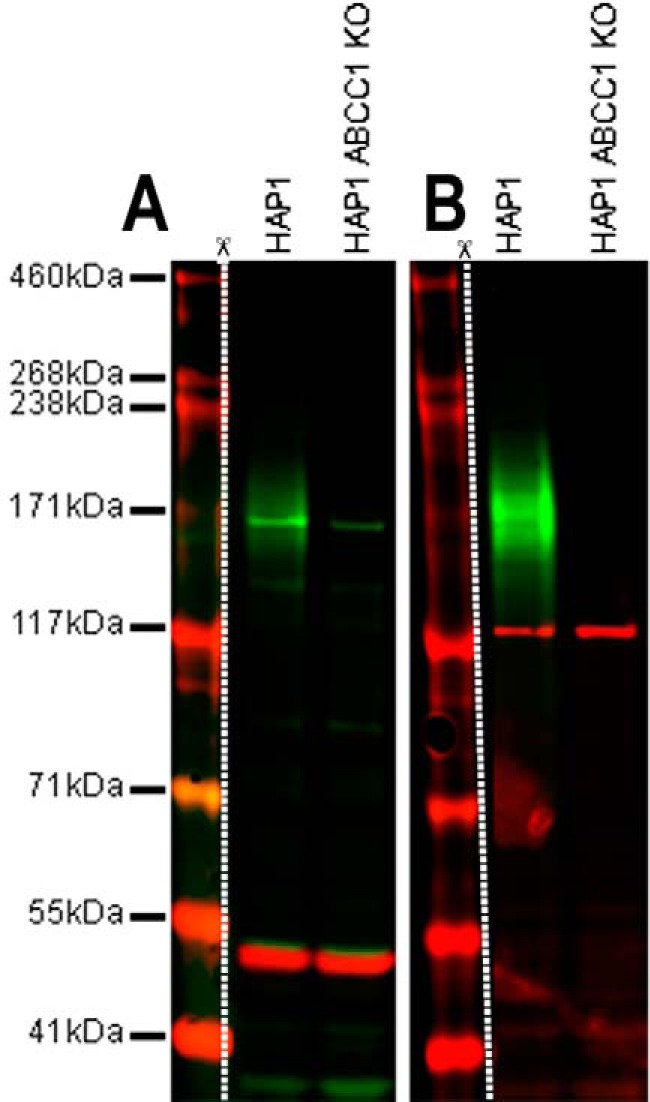
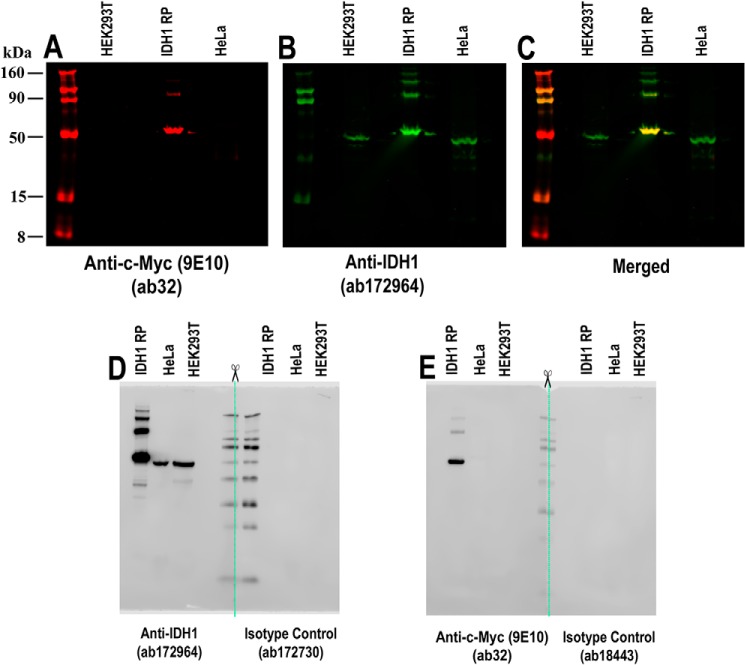
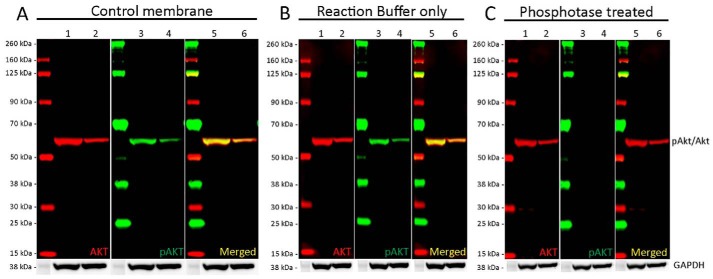
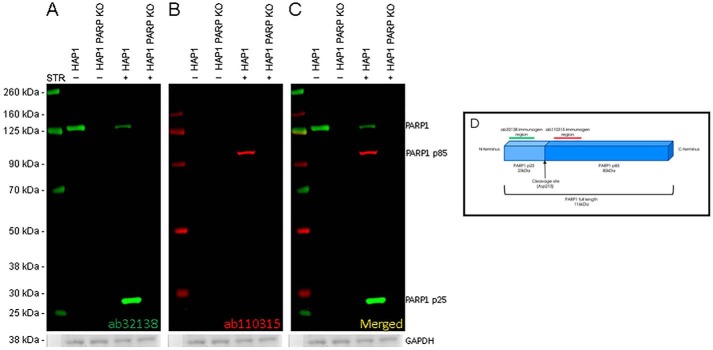
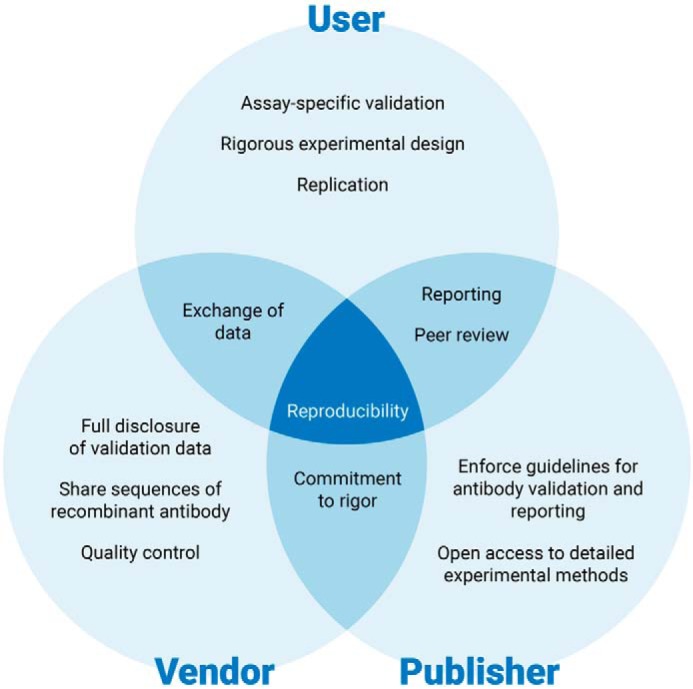
References
-
- Global Biological Standards Institute (2013) The case for standards in life science research. GBSI, Washington, D. C.
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
