

Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation
- PMID: 31819256
- PMCID: PMC6923591
- DOI: 10.1038/s41590-019-0550-7
Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation
Abstract
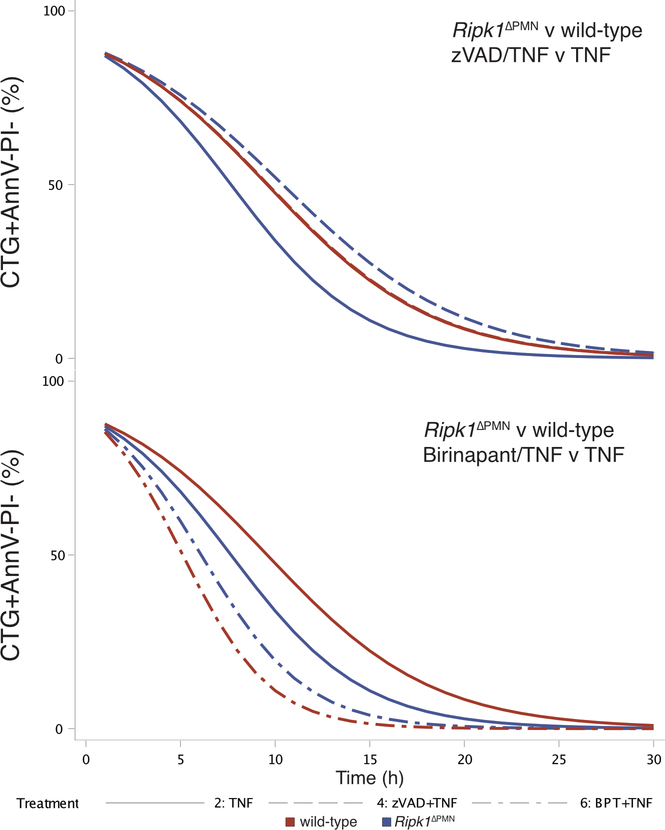
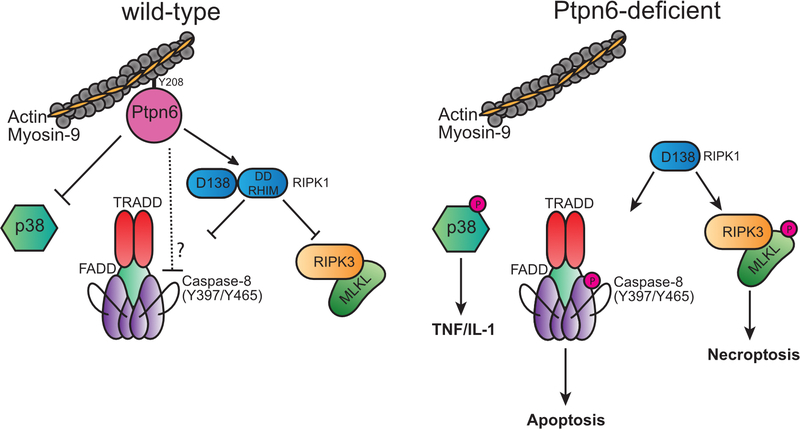
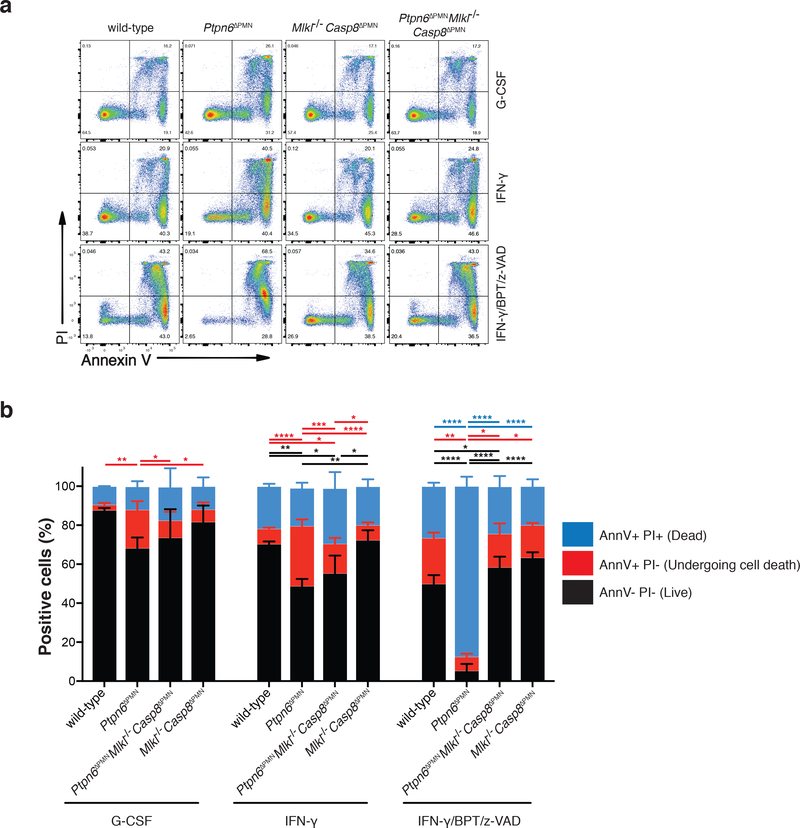
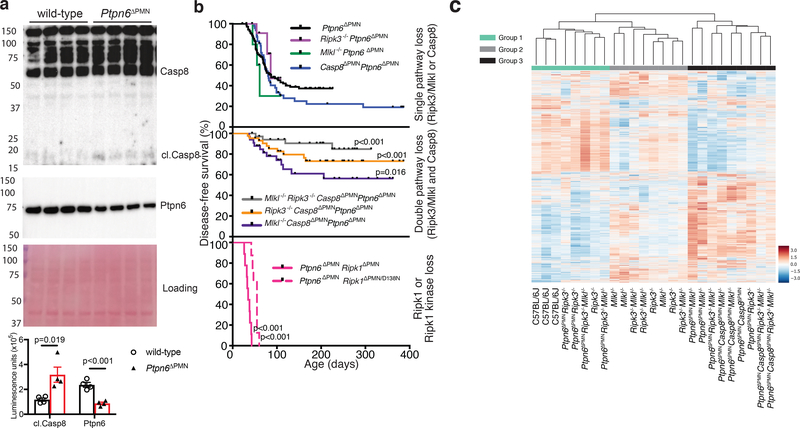
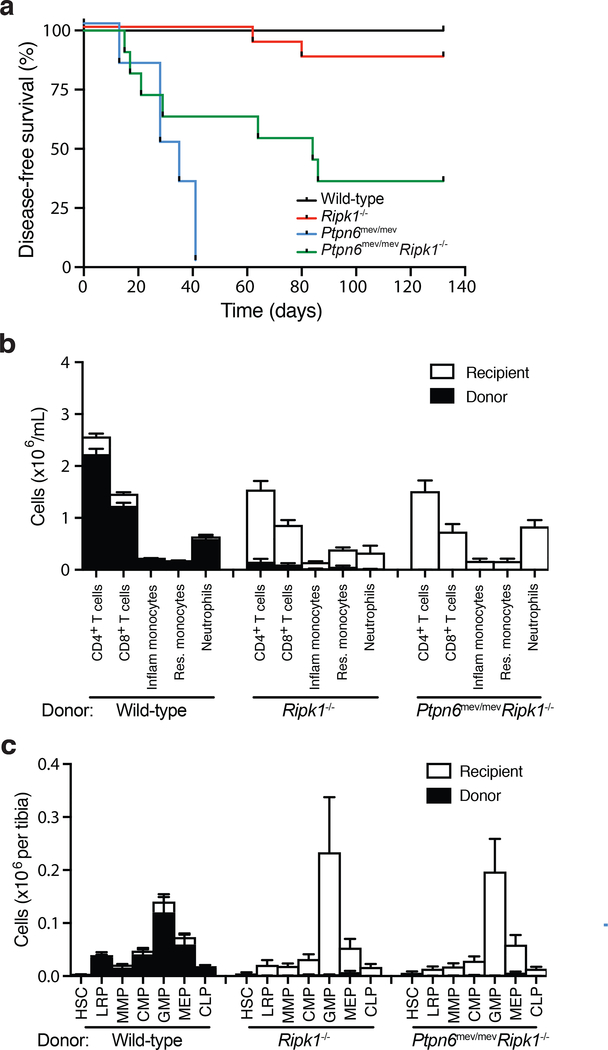
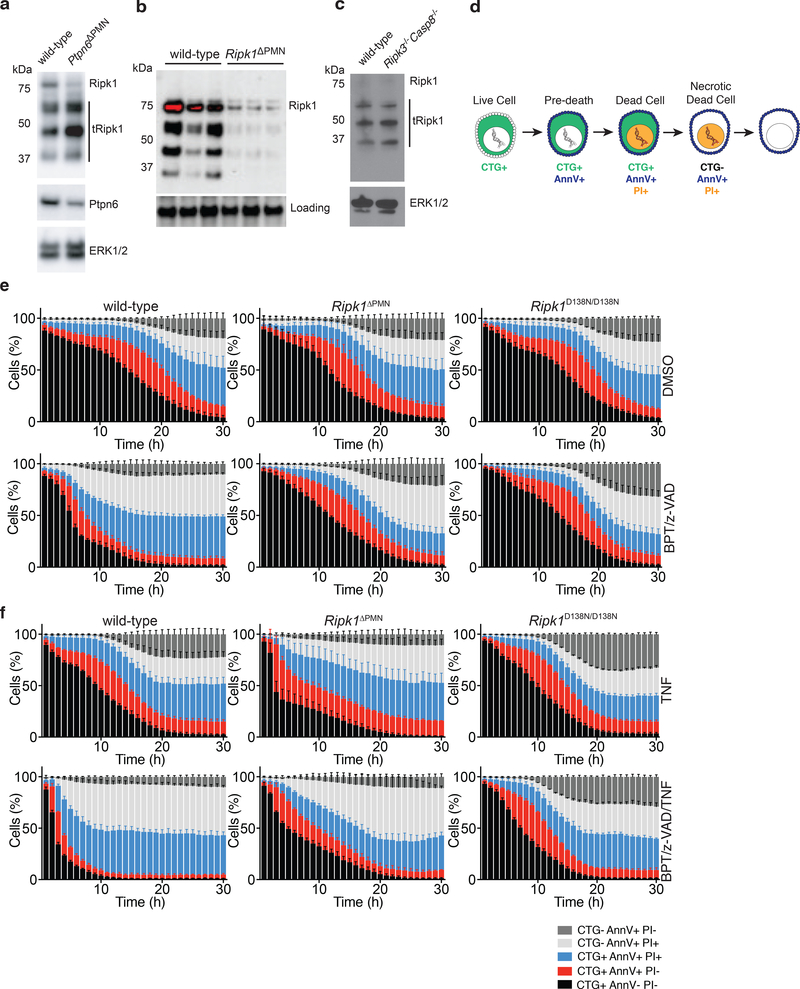
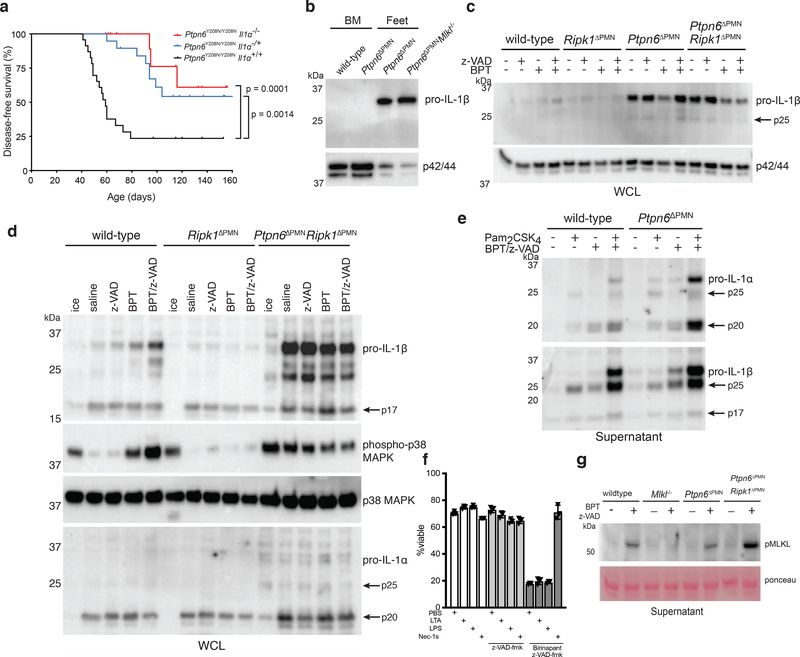
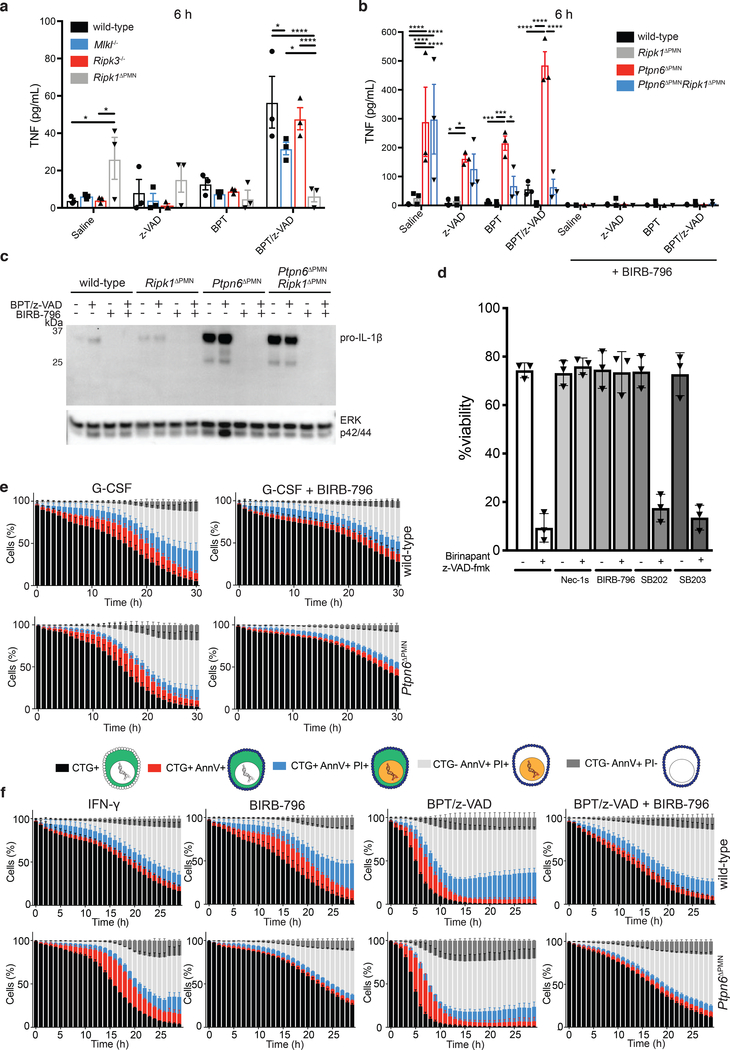
Ptpn6 is a cytoplasmic phosphatase that functions to prevent autoimmune and interleukin-1 (IL-1) receptor-dependent, caspase-1-independent inflammatory disease. Conditional deletion of Ptpn6 in neutrophils (Ptpn6∆PMN) is sufficient to initiate IL-1 receptor-dependent cutaneous inflammatory disease, but the source of IL-1 and the mechanisms behind IL-1 release remain unclear. Here, we investigate the mechanisms controlling IL-1α/β release from neutrophils by inhibiting caspase-8-dependent apoptosis and Ripk1-Ripk3-Mlkl-regulated necroptosis. Loss of Ripk1 accelerated disease onset, whereas combined deletion of caspase-8 and either Ripk3 or Mlkl strongly protected Ptpn6∆PMN mice. Ptpn6∆PMN neutrophils displayed increased p38 mitogen-activated protein kinase-dependent Ripk1-independent IL-1 and tumor necrosis factor production, and were prone to cell death. Together, these data emphasize dual functions for Ptpn6 in the negative regulation of p38 mitogen-activated protein kinase activation to control tumor necrosis factor and IL-1α/β expression, and in maintaining Ripk1 function to prevent caspase-8- and Ripk3-Mlkl-dependent cell death and concomitant IL-1α/β release.
Conflict of interest statement
Disclosure of Conflicts of Interest
The authors declare no competing financial or non-financial conflicts of interest.
Figures
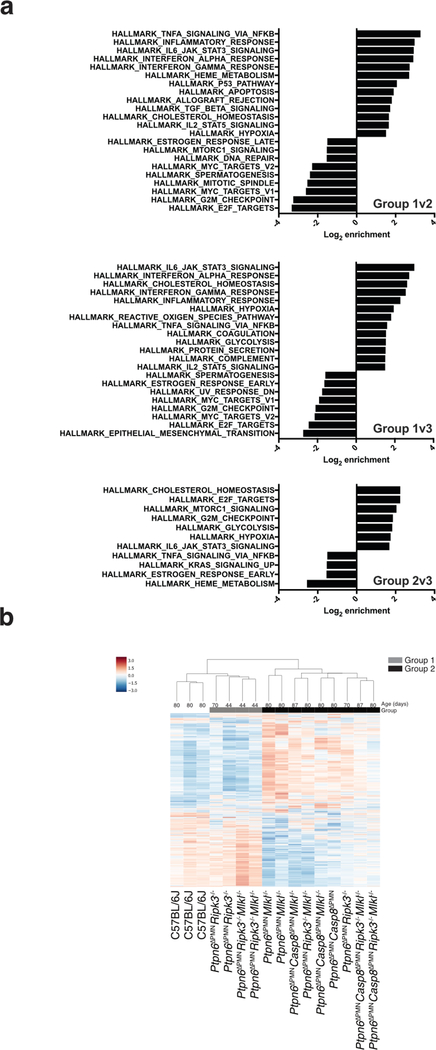
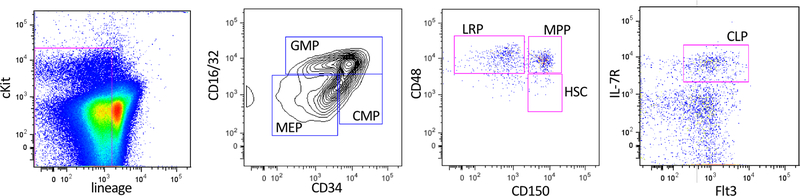
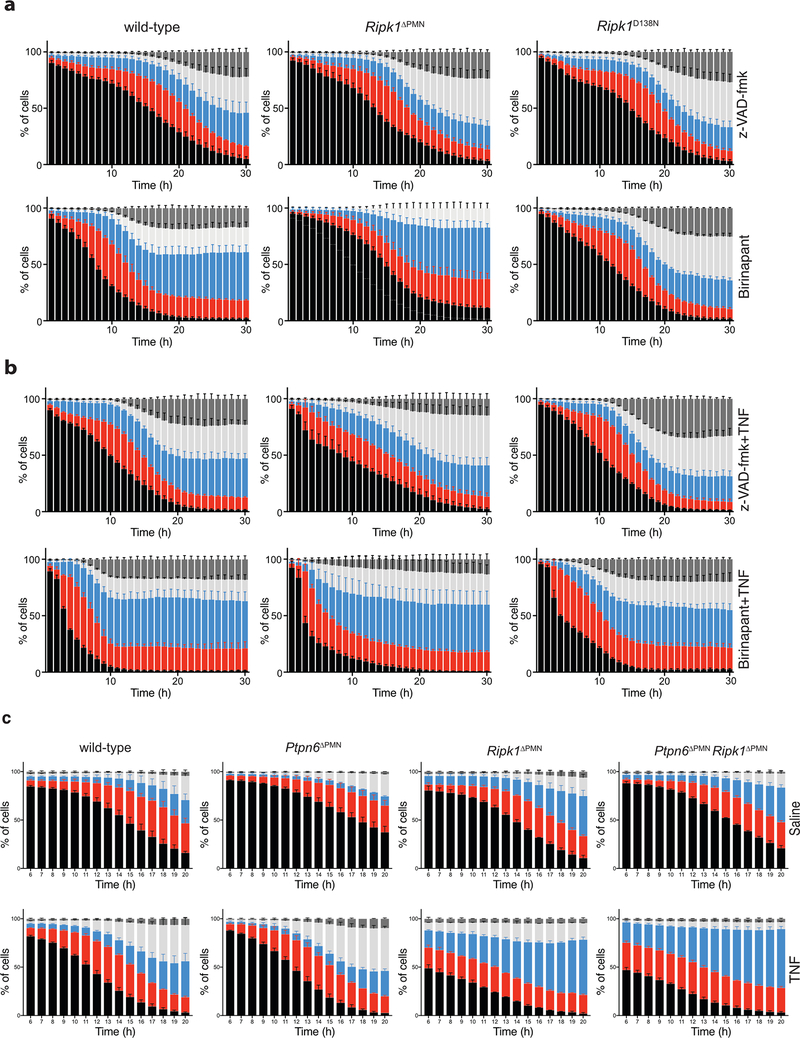
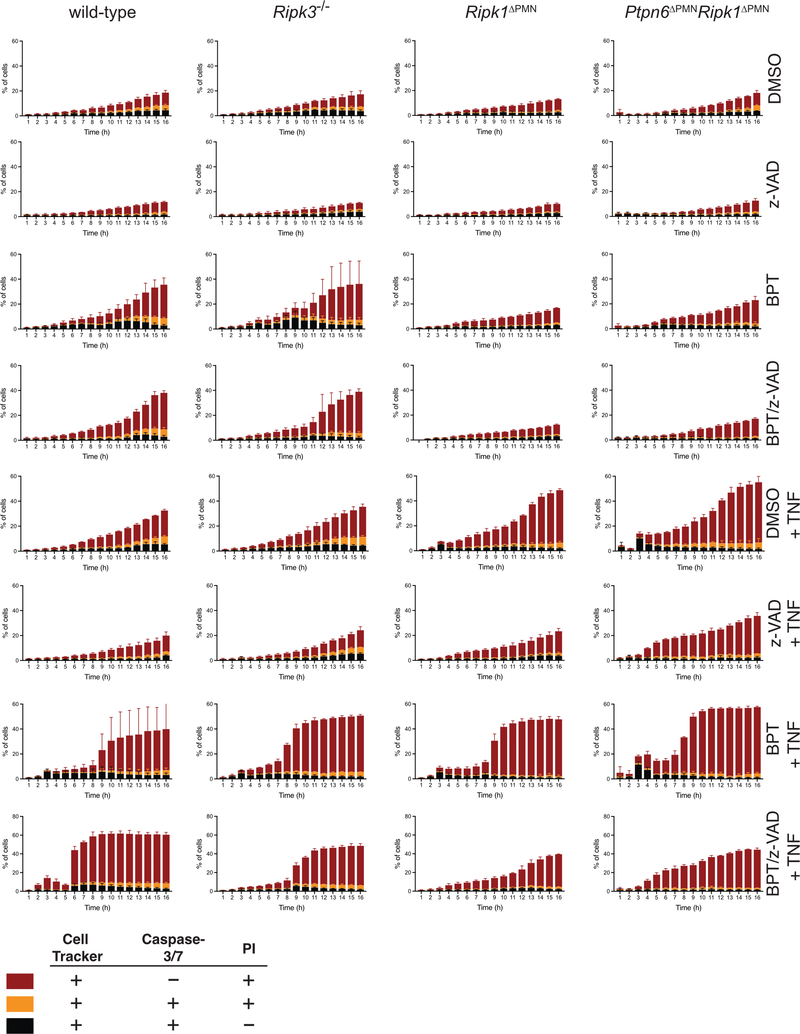
References
-
- Uihlein LC, Brandling-Bennett HA, Lio PA & Liang MG Sweet syndrome in children. Pediatr Dermatol 29, 38–44 (2012). - PubMed
-
- Prat L, Bouaziz JD, Wallach D, Vignon-Pennamen MD & Bagot M Neutrophilic dermatoses as systemic diseases. Clin Dermatol 32, 376–388 (2014). - PubMed
-
- Bourke JF, Jones JL, Fletcher A & Graham-Brown RA An immunohistochemical study of the dermal infiltrate and epidermal staining for interleukin 1 in 12 cases of Sweet’s syndrome. Br J Dermatol 134, 705–709 (1996). - PubMed
-
- Giasuddin AS, El-Orfi AH, Ziu MM & El-Barnawi NY Sweet’s syndrome: is the pathogenesis mediated by helper T cell type 1 cytokines? J Am Acad Dermatol 39, 940–943 (1998). - PubMed
METHODS-ONLY REFERENCES
-
- Pao LI et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity 27, 35–48 (2007). - PubMed
-
- Kelliher MA et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 8, 297–303 (1998). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
