

Directed evolution improves the catalytic efficiency of TEV protease
- PMID: 31819267
- PMCID: PMC7004888
- DOI: 10.1038/s41592-019-0665-7
Directed evolution improves the catalytic efficiency of TEV protease
Erratum in
-
Publisher Correction: Directed evolution improves the catalytic efficiency of TEV protease.Nat Methods. 2020 Feb;17(2):242. doi: 10.1038/s41592-019-0729-8. Nat Methods. 2020. PMID: 31907448
Abstract
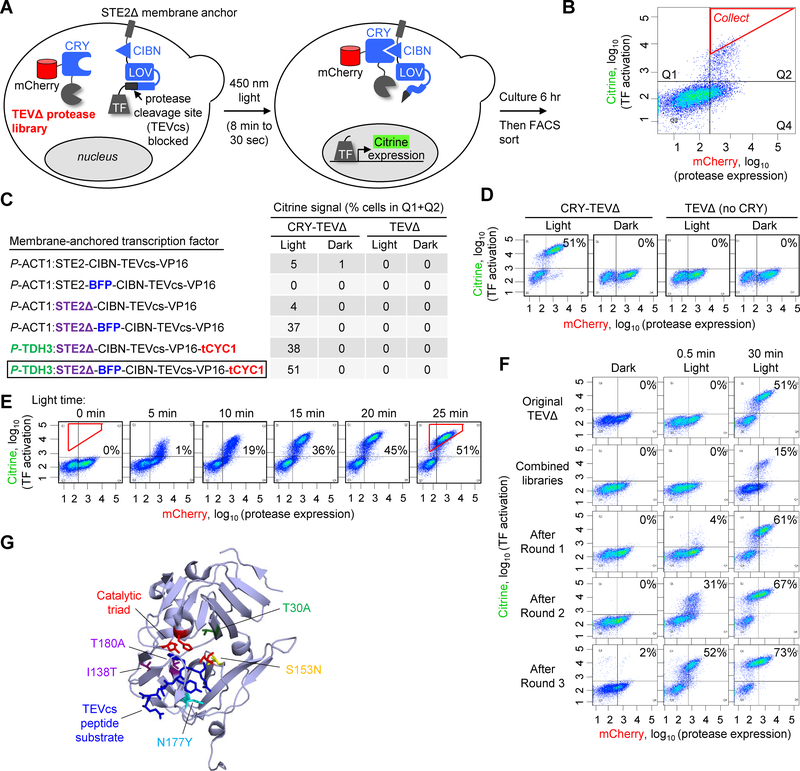
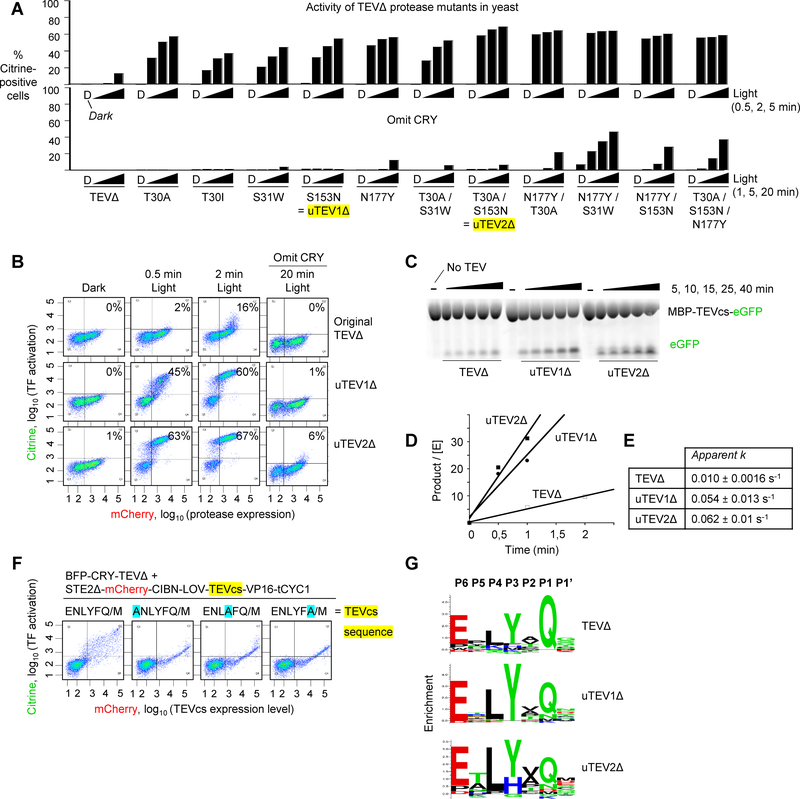
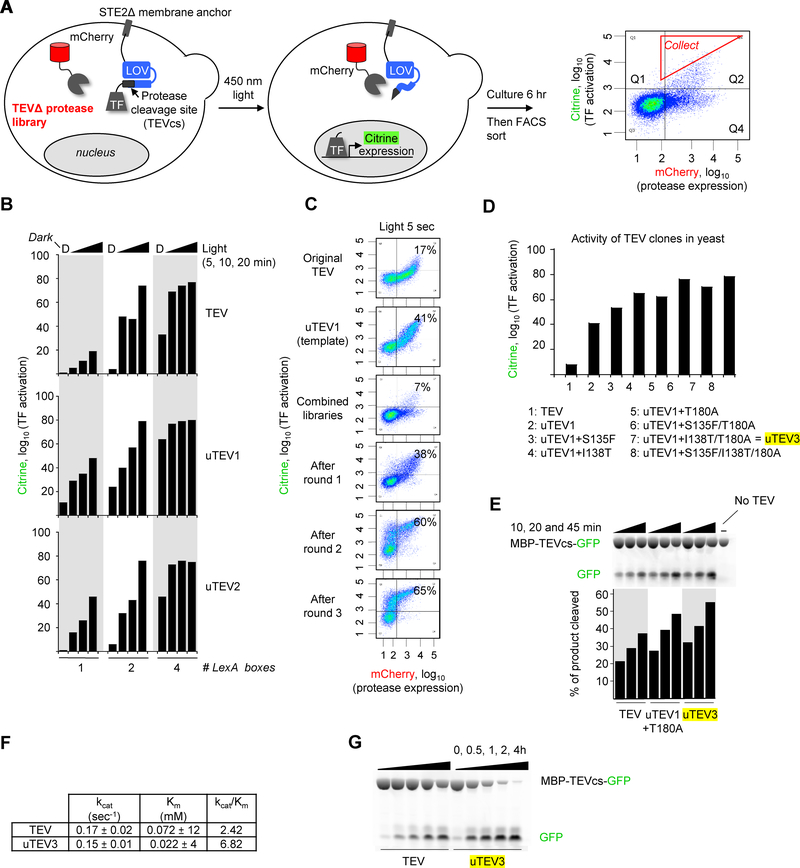
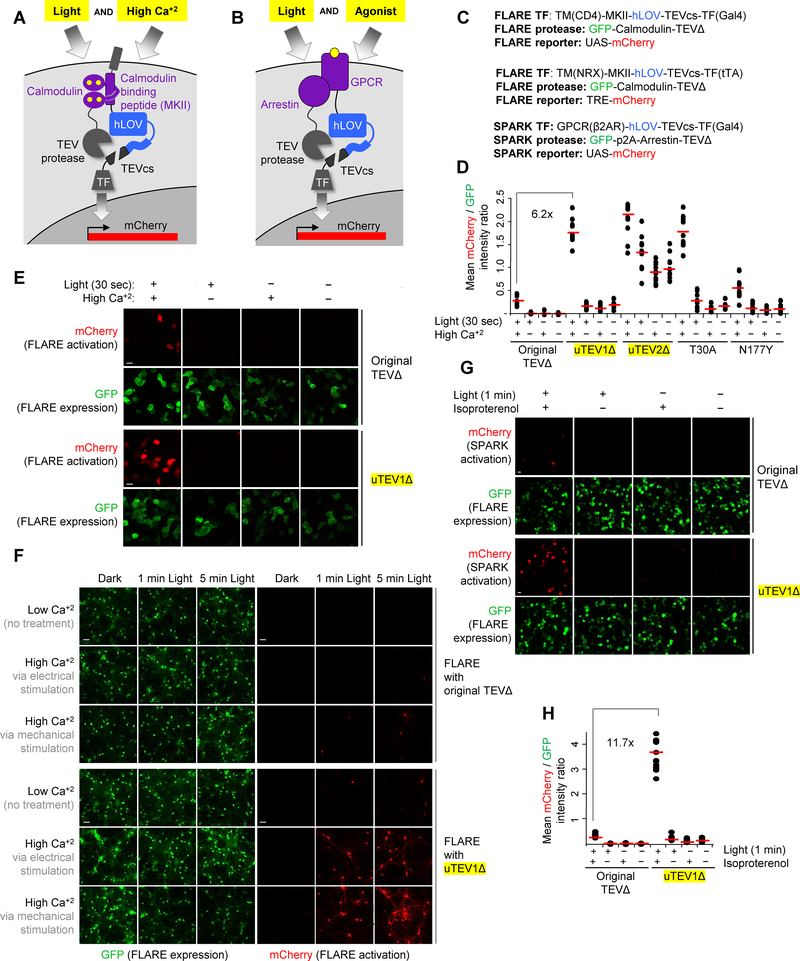
Tobacco etch virus protease (TEV) is one of the most widely used proteases in biotechnology because of its exquisite sequence specificity. A limitation, however, is its slow catalytic rate. We developed a generalizable yeast-based platform for directed evolution of protease catalytic properties. Protease activity is read out via proteolytic release of a membrane-anchored transcription factor, and we temporally regulate access to TEV's cleavage substrate using a photosensory LOV domain. By gradually decreasing light exposure time, we enriched faster variants of TEV over multiple rounds of selection. Our TEV-S153N mutant (uTEV1Δ), when incorporated into the calcium integrator FLARE, improved the signal/background ratio by 27-fold, and enabled recording of neuronal activity in culture with 60-s temporal resolution. Given the widespread use of TEV in biotechnology, both our evolved TEV mutants and the directed-evolution platform used to generate them could be beneficial across a wide range of applications.
Conflict of interest statement
COMPETING INTEREST
A.Y.T. and M.I.S have filed a patent application covering some aspects of this work.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
