

CPSF3-dependent pre-mRNA processing as a druggable node in AML and Ewing's sarcoma
- PMID: 31819276
- PMCID: PMC7116157
- DOI: 10.1038/s41589-019-0424-1
CPSF3-dependent pre-mRNA processing as a druggable node in AML and Ewing's sarcoma
Erratum in
-
Author Correction: CPSF3-dependent pre-mRNA processing as a druggable node in AML and Ewing's sarcoma.Nat Chem Biol. 2020 Apr;16(4):479. doi: 10.1038/s41589-020-0508-y. Nat Chem Biol. 2020. PMID: 32139909
Abstract
The post-genomic era has seen many advances in our understanding of cancer pathways, yet resistance and tumor heterogeneity necessitate multiple approaches to target even monogenic tumors. Here, we combine phenotypic screening with chemical genetics to identify pre-messenger RNA endonuclease cleavage and polyadenylation specificity factor 3 (CPSF3) as the target of JTE-607, a small molecule with previously unknown target. We show that CPSF3 represents a synthetic lethal node in a subset of acute myeloid leukemia (AML) and Ewing's sarcoma cancer cell lines. Inhibition of CPSF3 by JTE-607 alters expression of known downstream effectors in AML and Ewing's sarcoma lines, upregulates apoptosis and causes tumor-selective stasis in mouse xenografts. Mechanistically, it prevents the release of newly synthesized pre-mRNAs, resulting in read-through transcription and the formation of DNA-RNA hybrid R-loop structures. This study implicates pre-mRNA processing, and specifically CPSF3, as a druggable target providing an avenue to therapeutic intervention in cancer.
Conflict of interest statement
All authors (except otherwise noted) were employees of Novartis Institutes for BioMedical Research at the time of their involvement in this study and may hold stock in Novartis. Correspondence should be addressed to R.E.J.B. (
Figures
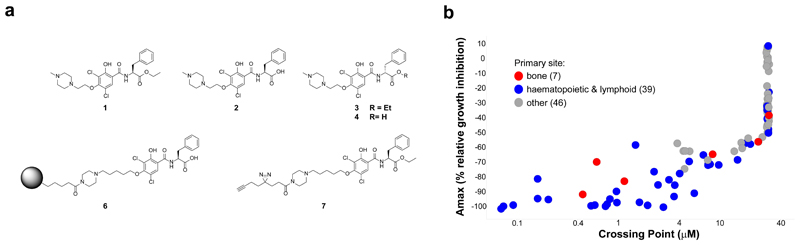
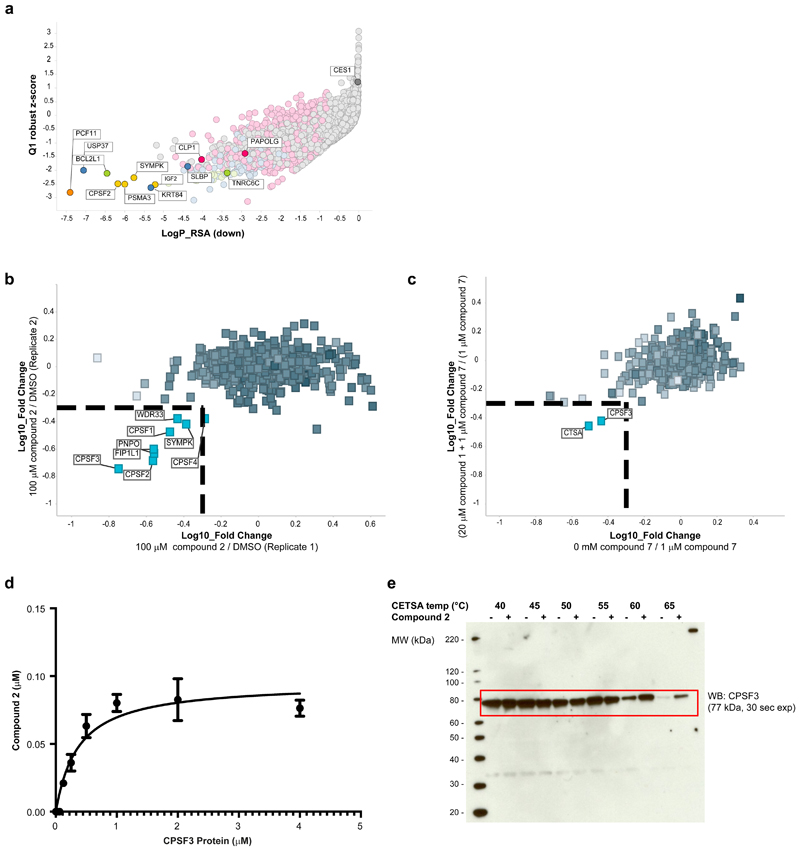
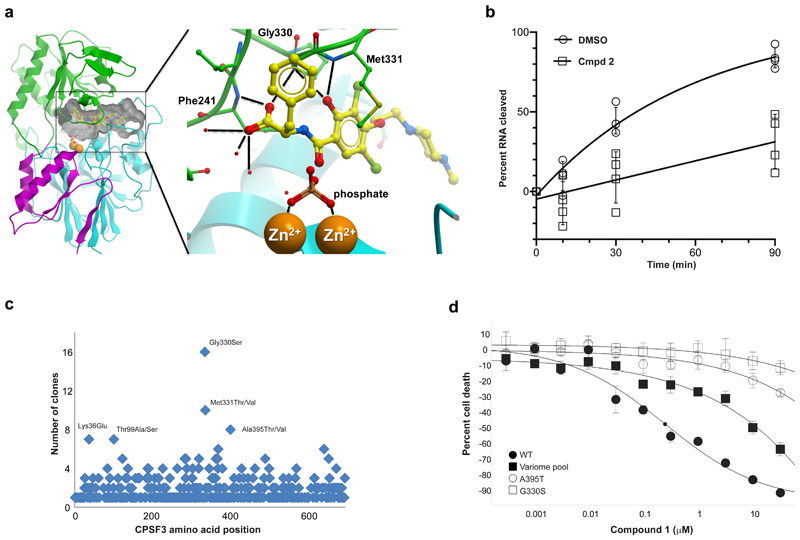
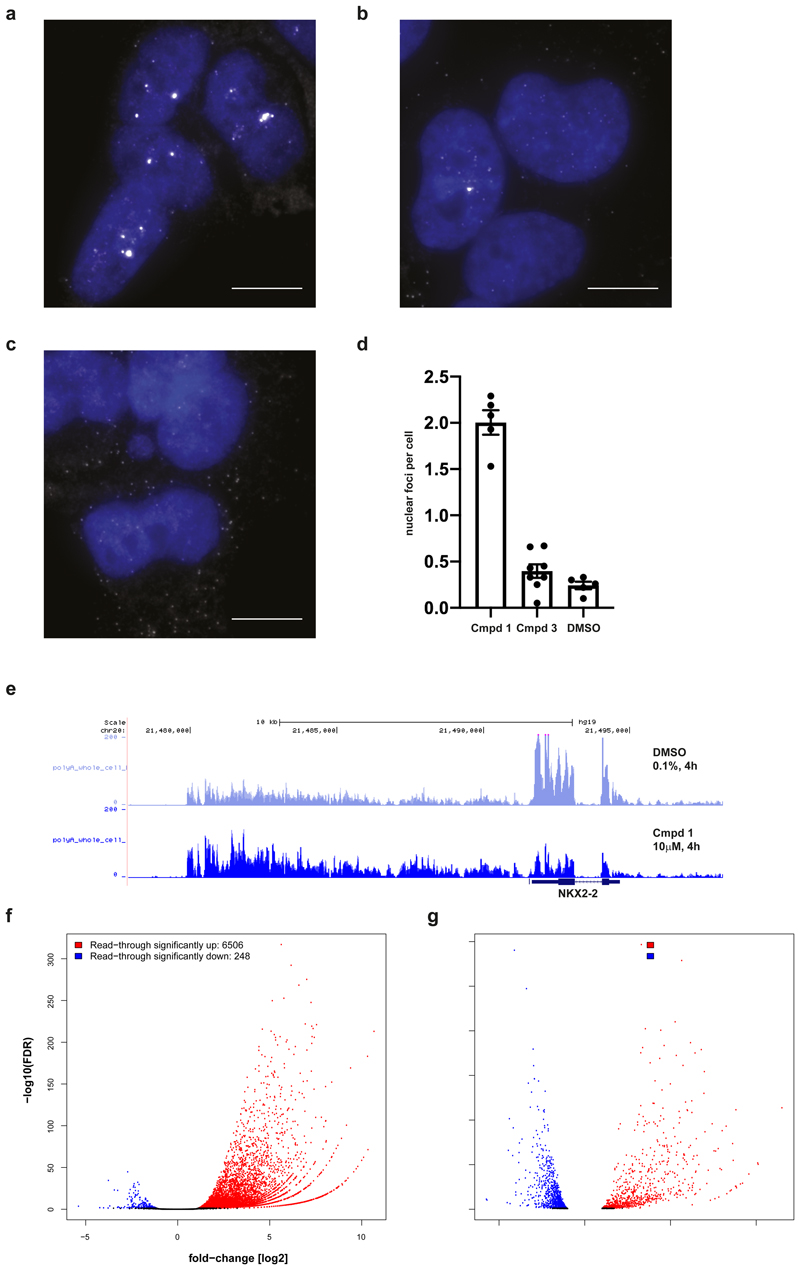
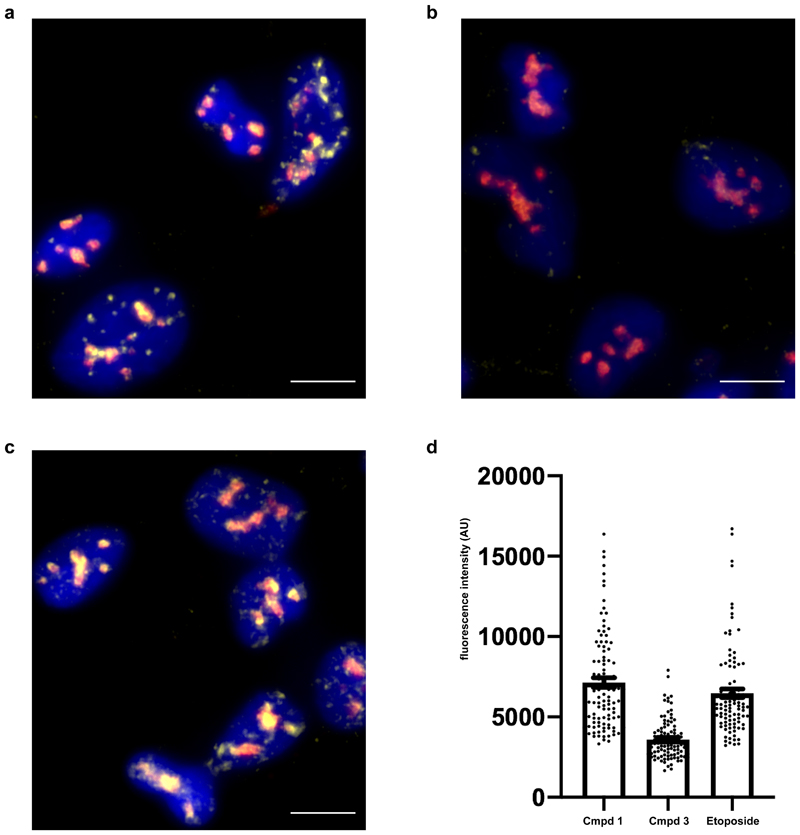
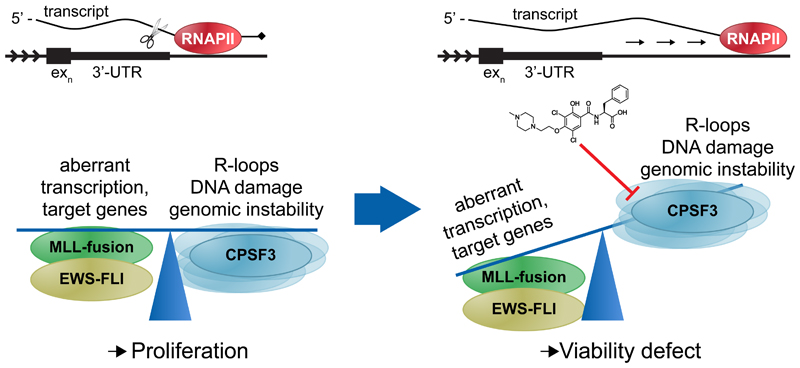
Comment in
-
Processing for destruction.Nat Chem Biol. 2020 Jan;16(1):3-4. doi: 10.1038/s41589-019-0428-x. Nat Chem Biol. 2020. PMID: 31819275 No abstract available.
References
-
- Swinney DC, Anthony J. How were new medicines discovered? Nat. Rev. Drug Discov. 2011;10:507–19. - PubMed
-
- Moffat JG, Vincent F, Lee JA, Eder J, Prunotto M. Opportunities and challenges in phenotypic drug discovery: An industry perspective. Nat. Rev. Drug Discov. 2017;16:531–543. - PubMed
-
- Schreiber SL. Chemical genetics resulting from a passion for synthetic organic chemistry. Bioorg. Med. Chem. 1998;6:1127–1152. - PubMed
-
- Rothman DM, et al. Metabolic Enzyme Sulfotransferase 1A1 Is the Trigger for N-Benzyl Indole Carbinol Tumor Growth Suppression. Chem. Biol. 2015;22:1228–1237. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
