

Direct electrophysiological evidence that spreading depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura and a review of the spreading depolarization continuum of acute neuronal mass injury
- PMID: 31820363
- PMCID: PMC7031471
- DOI: 10.1007/s11357-019-00142-7
Direct electrophysiological evidence that spreading depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura and a review of the spreading depolarization continuum of acute neuronal mass injury
Abstract
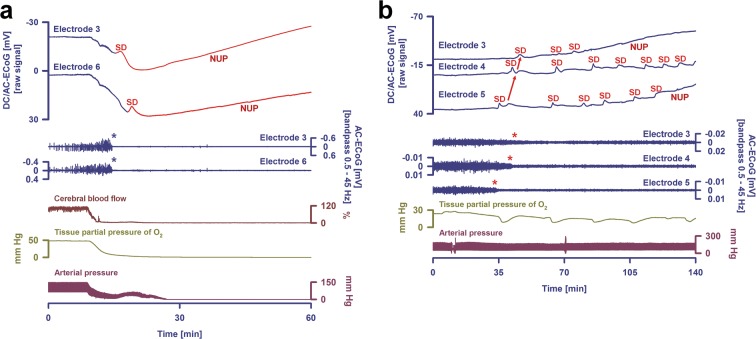
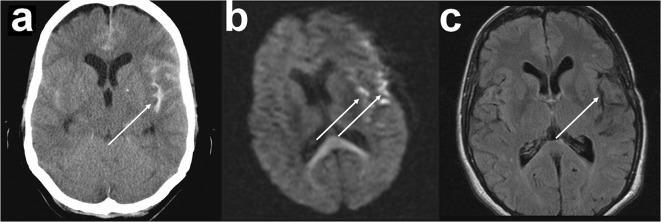
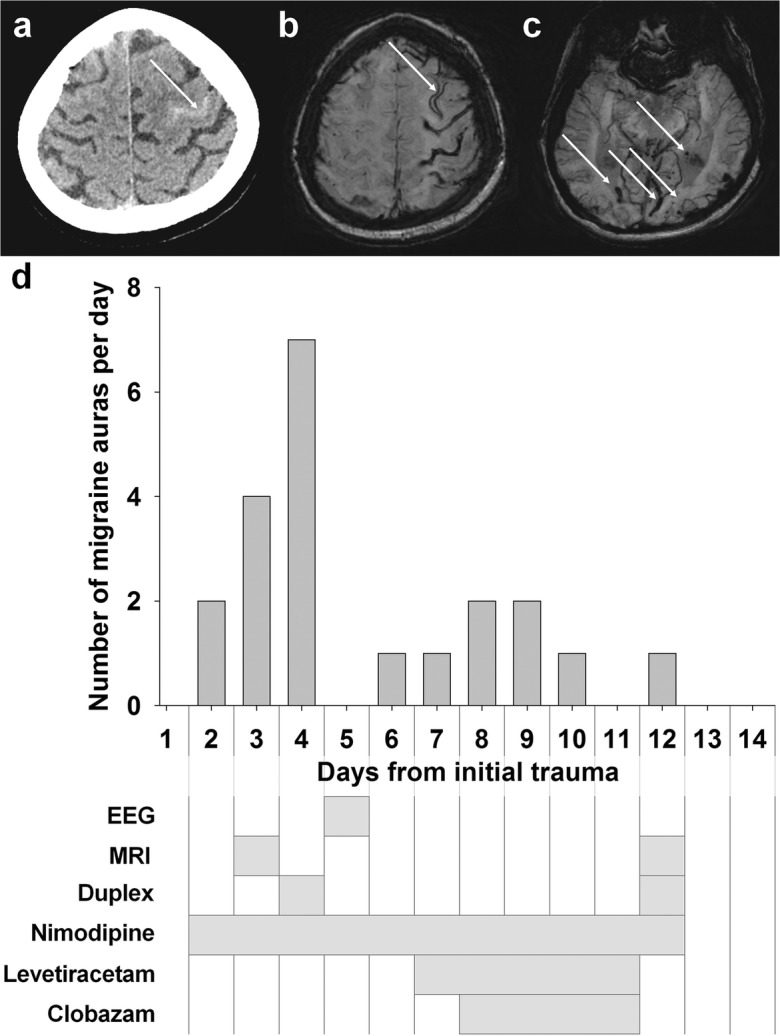
Spreading depolarization is observed as a large negative shift of the direct current potential, swelling of neuronal somas, and dendritic beading in the brain's gray matter and represents a state of a potentially reversible mass injury. Its hallmark is the abrupt, massive ion translocation between intraneuronal and extracellular compartment that causes water uptake (= cytotoxic edema) and massive glutamate release. Dependent on the tissue's energy status, spreading depolarization can co-occur with different depression or silencing patterns of spontaneous activity. In adequately supplied tissue, spreading depolarization induces spreading depression of activity. In severely ischemic tissue, nonspreading depression of activity precedes spreading depolarization. The depression pattern determines the neurological deficit which is either spreading such as in migraine aura or migraine stroke or nonspreading such as in transient ischemic attack or typical stroke. Although a clinical distinction between spreading and nonspreading focal neurological deficits is useful because they are associated with different probabilities of permanent damage, it is important to note that spreading depolarization, the neuronal injury potential, occurs in all of these conditions. Here, we first review the scientific basis of the continuum of spreading depolarizations. Second, we highlight the transition zone of the continuum from reversibility to irreversibility using clinical cases of aneurysmal subarachnoid hemorrhage and cerebral amyloid angiopathy. These illustrate how modern neuroimaging and neuromonitoring technologies increasingly bridge the gap between basic sciences and clinic. For example, we provide direct electrophysiological evidence for the first time that spreading depolarization-induced spreading depression is the pathophysiological correlate of the migraine aura.
Keywords: Cerebral amyloid angiopathy; Delayed cerebral ischemia; Migraine aura; Spreading depression; Subarachnoid hemorrhage.
Figures


Similar articles
-
Migraine Aura, Transient Ischemic Attacks, Stroke, and Dying of the Brain Share the Same Key Pathophysiological Process in Neurons Driven by Gibbs-Donnan Forces, Namely Spreading Depolarization.Front Cell Neurosci. 2022 Feb 10;16:837650. doi: 10.3389/fncel.2022.837650. eCollection 2022. Front Cell Neurosci. 2022. PMID: 35237133 Free PMC article. Review.
-
How spreading depolarization can be the pathophysiological correlate of both migraine aura and stroke.Acta Neurochir Suppl. 2015;120:137-40. doi: 10.1007/978-3-319-04981-6_23. Acta Neurochir Suppl. 2015. PMID: 25366613
-
Chloride Cotransporters as a Molecular Mechanism underlying Spreading Depolarization-Induced Dendritic Beading.J Neurosci. 2015 Sep 2;35(35):12172-87. doi: 10.1523/JNEUROSCI.0400-15.2015. J Neurosci. 2015. PMID: 26338328 Free PMC article.
-
The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease.Nat Med. 2011 Apr;17(4):439-47. doi: 10.1038/nm.2333. Epub 2011 Apr 7. Nat Med. 2011. PMID: 21475241 Review.
-
Recording, analysis, and interpretation of spreading depolarizations in neurointensive care: Review and recommendations of the COSBID research group.J Cereb Blood Flow Metab. 2017 May;37(5):1595-1625. doi: 10.1177/0271678X16654496. Epub 2016 Jan 1. J Cereb Blood Flow Metab. 2017. PMID: 27317657 Free PMC article. Review.
Cited by
-
Stroke-prone salt-sensitive spontaneously hypertensive rats show higher susceptibility to spreading depolarization (SD) and altered hemodynamic responses to SD.J Cereb Blood Flow Metab. 2023 Feb;43(2):210-230. doi: 10.1177/0271678X221135085. Epub 2022 Nov 3. J Cereb Blood Flow Metab. 2023. PMID: 36329390 Free PMC article.
-
Lasting s-ketamine block of spreading depolarizations in subarachnoid hemorrhage: a retrospective cohort study.Crit Care. 2019 Dec 30;23(1):427. doi: 10.1186/s13054-019-2711-3. Crit Care. 2019. PMID: 31888772 Free PMC article.
-
Initiation of migraine-related cortical spreading depolarization by hyperactivity of GABAergic neurons and NaV1.1 channels.J Clin Invest. 2021 Nov 1;131(21):e142203. doi: 10.1172/JCI142203. J Clin Invest. 2021. PMID: 34491914 Free PMC article.
-
Narrative review of roles of astrocytes in subarachnoid hemorrhage.Ann Transl Med. 2023 Apr 28;11(8):302. doi: 10.21037/atm-22-5486. Epub 2023 Mar 23. Ann Transl Med. 2023. PMID: 37181334 Free PMC article. Review.
-
Quantifying the relationship between spreading depolarization and perivascular cerebrospinal fluid flow.Sci Rep. 2023 Jul 31;13(1):12405. doi: 10.1038/s41598-023-38938-5. Sci Rep. 2023. PMID: 37524734 Free PMC article.
References
-
- Aitken PG, Tombaugh GC, Turner DA, Somjen GG. Similar propagation of SD and hypoxic SD-like depolarization in rat hippocampus recorded optically and electrically. J Neurophysiol. 1998;80:1514–1521. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
