

Association of CMV genomic mutations with symptomatic infection and hearing loss in congenital CMV infection
- PMID: 31822287
- PMCID: PMC6905059
- DOI: 10.1186/s12879-019-4681-0
Association of CMV genomic mutations with symptomatic infection and hearing loss in congenital CMV infection
Erratum in
-
Correction to: Association of CMV genomic mutations with symptomatic infection and hearing loss in congenital CMV infection.BMC Infect Dis. 2020 Feb 10;20(1):111. doi: 10.1186/s12879-020-4766-9. BMC Infect Dis. 2020. PMID: 32039707 Free PMC article.
Abstract
Background: Congenital cytomegalovirus (cCMV) infection is the most common congenital infection and a leading cause of long-term neurological and sensory sequelae, the most common being sensorineural hearing loss (SNHL). Despite extensive research, clinical or laboratory markers to identify CMV infected children with increased risk for disease have not been identified. This study utilizes viral whole-genome next generation-sequencing (NGS) of specimens from congenitally infected infants to explore viral diversity and specific viral variants that may be associated with symptomatic infection and SNHL.
Methods: CMV DNA from urine specimens of 30 infants (17 asymptomatic, 13 symptomatic) was target enriched and next generation sequenced resulting in 93% coverage of the CMV genome allowing analysis of viral diversity.
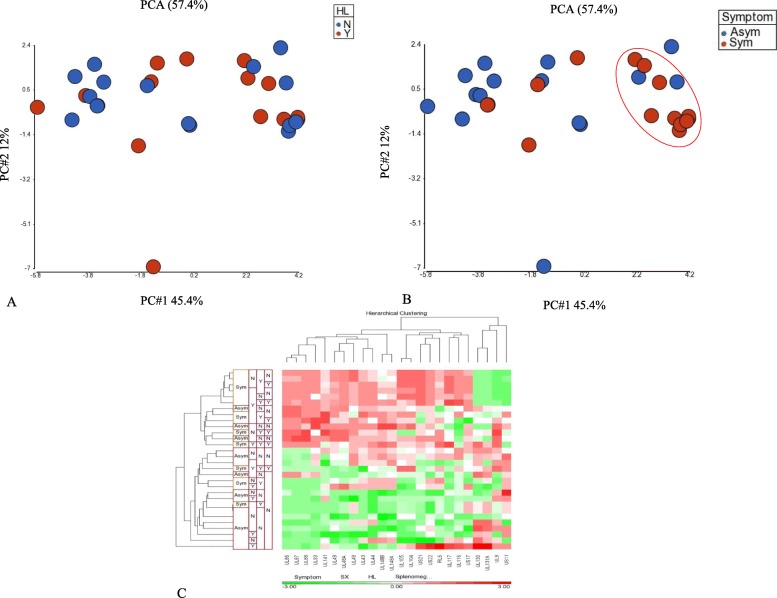
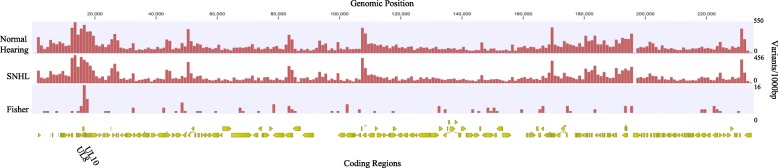
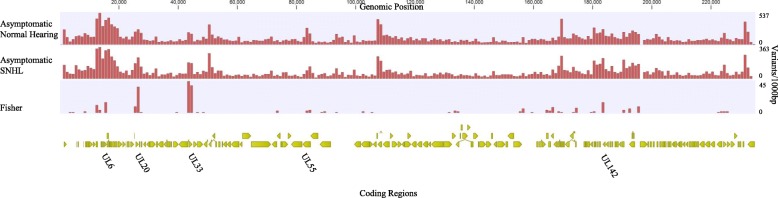
Results: Variant frequency distribution was compared between children with symptomatic and asymptomatic cCMV and those with (n = 13) and without (n = 17) hearing loss. The CMV genes UL48A, UL88, US19 and US22 were found to have an increase in nucleotide diversity in symptomatic children; while UL57, UL20, UL104, US14, UL115, and UL35 had an increase in diversity in children with hearing loss. An analysis of single variant differences between symptomatic and asymptomatic children found UL55 to have the highest number, while the most variants associated with SNHL were in the RL11 gene family. In asymptomatic infants with SNHL, mutations were observed more frequently in UL33 and UL20.
Conclusion: CMV genomes from infected newborns can be mapped to 93% of the genome at a depth allowing accurate and reproducible analysis of polymorphisms for variant and gene discovery that may be linked to symptomatic and hearing loss outcomes.
Keywords: Cytomegalovirus (CMV); Next generation sequencing (NGS); Sensory neural hearing loss (SNHL); Viral diversity.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
-
- Britt W. Cytomegalovirus. In: Remington JS, Klein JO, Wilson CB, Baker CJ, editors. Infectious Diseases of the Fetus and Newborn Infant. 7. Philadelphia: W.B. Saunders Company; 2011. pp. 704–753.
-
- Sijmons Steven, Thys Kim, Mbong Ngwese Mirabeau, Van Damme Ellen, Dvorak Jan, Van Loock Marnix, Li Guangdi, Tachezy Ruth, Busson Laurent, Aerssens Jeroen, Van Ranst Marc, Maes Piet. High-Throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-Disrupting Mutations and Pervasive Recombination. Journal of Virology. 2015;89(15):7673–7695. doi: 10.1128/JVI.00578-15. - DOI - PMC - PubMed
