

Transcriptional correlates of the pathological phenotype in a Huntington's disease mouse model
- PMID: 31822756
- PMCID: PMC6904489
- DOI: 10.1038/s41598-019-55177-9
Transcriptional correlates of the pathological phenotype in a Huntington's disease mouse model
Abstract
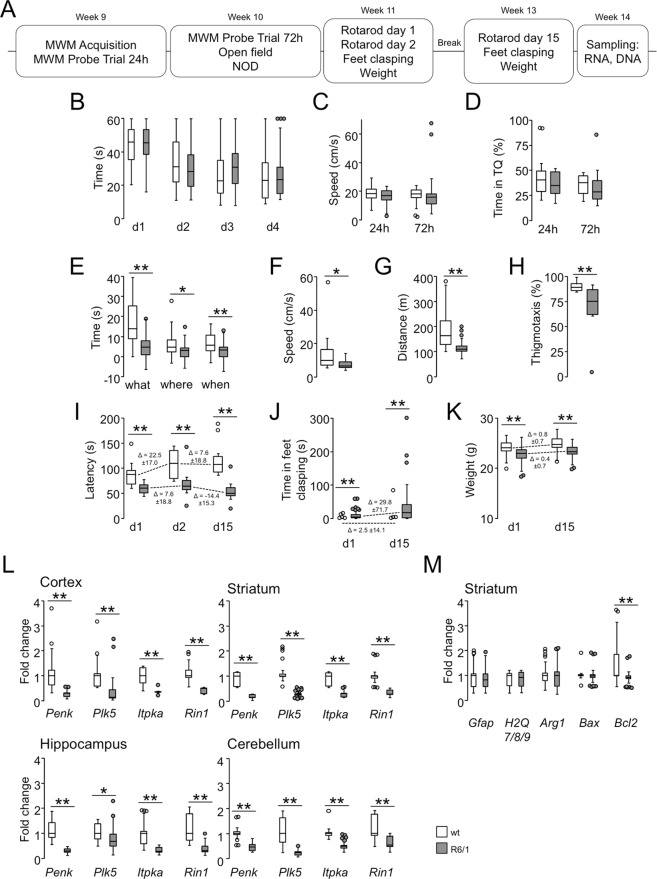
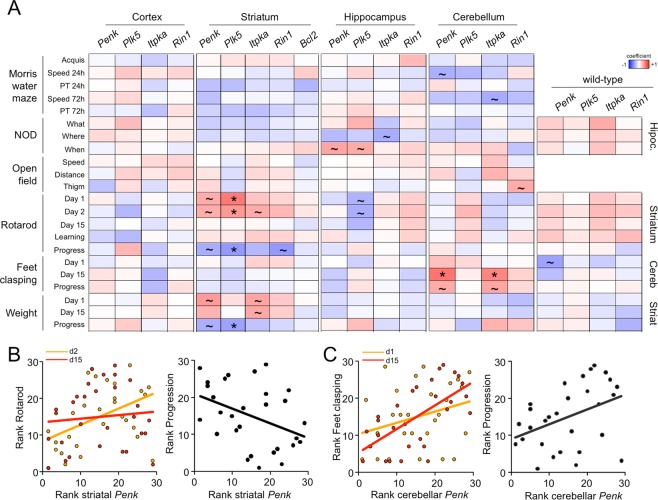
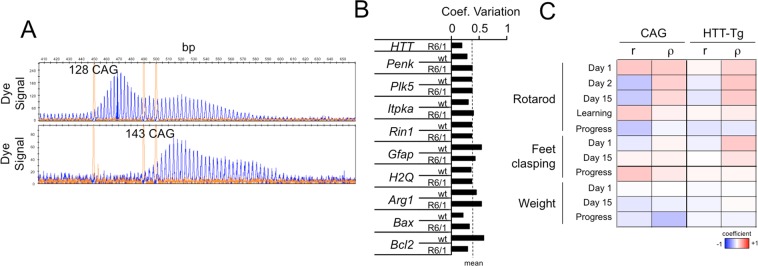
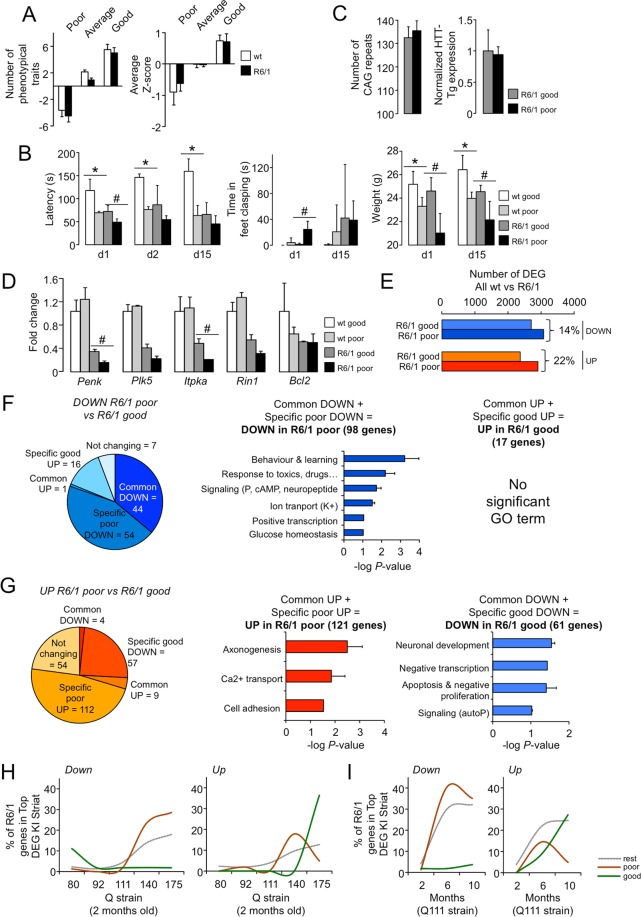
Huntington disease (HD) is a fatal neurodegenerative disorder without a cure that is caused by an aberrant expansion of CAG repeats in exon 1 of the huntingtin (HTT) gene. Although a negative correlation between the number of CAG repeats and the age of disease onset is established, additional factors may contribute to the high heterogeneity of the complex manifestation of symptoms among patients. This variability is also observed in mouse models, even under controlled genetic and environmental conditions. To better understand this phenomenon, we analysed the R6/1 strain in search of potential correlates between pathological motor/cognitive phenotypical traits and transcriptional alterations. HD-related genes (e.g., Penk, Plk5, Itpka), despite being downregulated across the examined brain areas (the prefrontal cortex, striatum, hippocampus and cerebellum), exhibited tissue-specific correlations with particular phenotypical traits that were attributable to the contribution of the brain region to that trait (e.g., striatum and rotarod performance, cerebellum and feet clasping). Focusing on the striatum, we determined that the transcriptional dysregulation associated with HD was partially exacerbated in mice that showed poor overall phenotypical scores, especially in genes with relevant roles in striatal functioning (e.g., Pde10a, Drd1, Drd2, Ppp1r1b). However, we also observed transcripts associated with relatively better outcomes, such as Nfya (CCAAT-binding transcription factor NF-Y subunit A) plus others related to neuronal development, apoptosis and differentiation. In this study, we demonstrated that altered brain transcription can be related to the manifestation of HD-like symptoms in mouse models and that this can be extrapolated to the highly heterogeneous population of HD patients.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
