

Targeted deep-intronic sequencing in a cohort of unexplained cases of suspected Lynch syndrome
- PMID: 31822864
- PMCID: PMC7170855
- DOI: 10.1038/s41431-019-0536-9
Targeted deep-intronic sequencing in a cohort of unexplained cases of suspected Lynch syndrome
Abstract
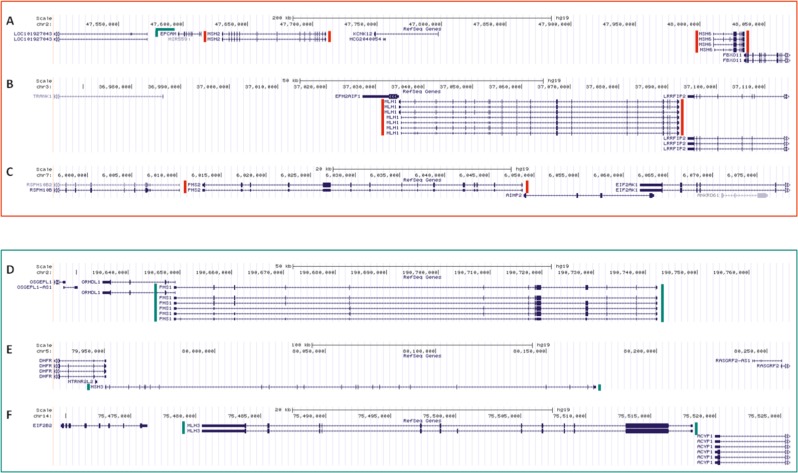
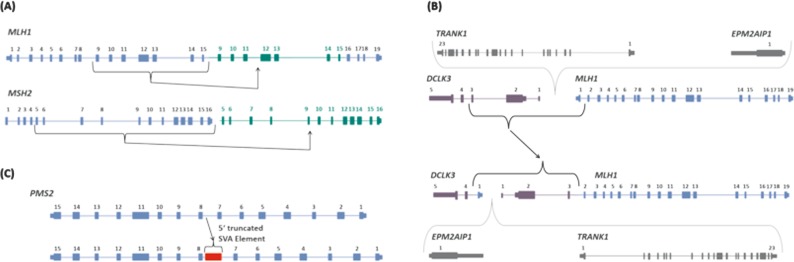
Lynch syndrome (LS) is caused by germline defects in DNA mismatch repair (MMR) pathway, resulting in microsatellite instability (MSI-H) and loss of immunohistochemical staining (IHC) of the respective protein in tumor tissue. However, not in all clinically suspected LS patients with MSI-H tumors and IHC-loss, causative germline alterations in the MMR genes can be detected. Here, we investigated 128 of these patients to possibly define new pathomechanisms. A search for large genomic rearrangements and deep-intronic regulatory variants was performed via targeted next-generation sequencing (NGS) of exonic, intronic, and chromosomal regions upstream and downstream of MLH1, MSH2, MSH6, PMS2, MLH3, MSH3, PMS1, and EPCAM. Within this cohort, two different large rearrangements causative for LS were detected in three cases, belonging to two families (2.3%). The sensitivity to detect large rearrangements or copy number variations (CNV) was evaluated to be 50%. In 9 of the 128 patients (7%), previously overlooked pathogenic single-nucleotide variants (SNV) and two variants of uncertain significance (VUS) were identified in MLH1, MSH2, and MSH6. Pathogenic aberrations were not found in MLH3, MSH3, and PMS1. A potential effect on regulation was exerted for 19% of deep-intronic SNVs, predominantly located in chromosomal regions where the modification of histone proteins suggests an enhancer function. In conclusion, conventional variation analysis of coding regions is missing rare genomic rearrangements, nevertheless they should be analyzed. Assessment of deep-intronic SNVs is so far non-conclusive for medical questioning.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


References
-
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. - PubMed
-
- Jass JR. What’s new in hereditary colorectal cancer? Arch Pathol Lab Med. 2005;129:1380–4. - PubMed
-
- Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer) N Engl J Med. 2005;352:1851–60.. - PubMed
-
- Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–32.. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
