

Clinical and Biochemical Phenotypes in a Family With ENPP1 Mutations
- PMID: 31826312
- PMCID: PMC7771569
- DOI: 10.1002/jbmr.3938
Clinical and Biochemical Phenotypes in a Family With ENPP1 Mutations
Abstract
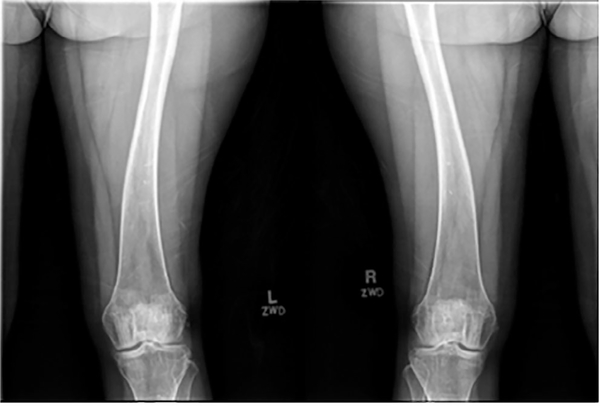
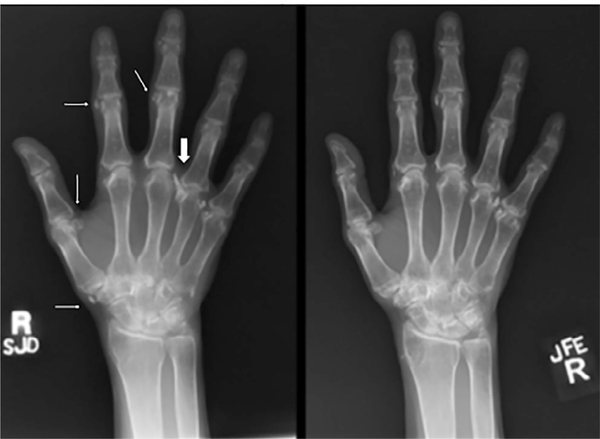
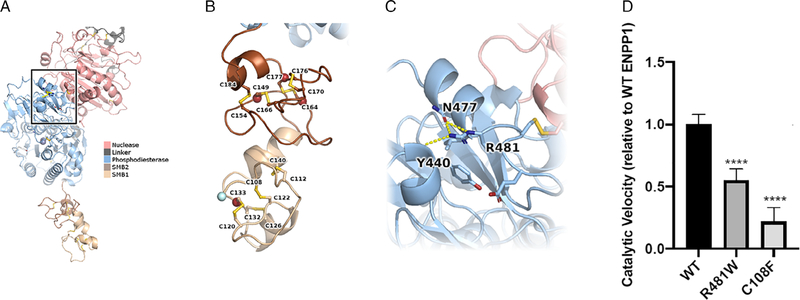
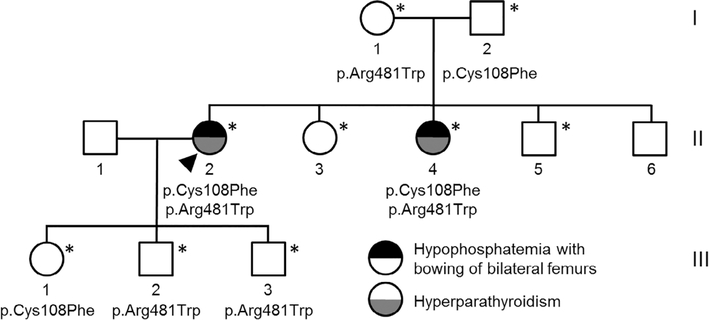
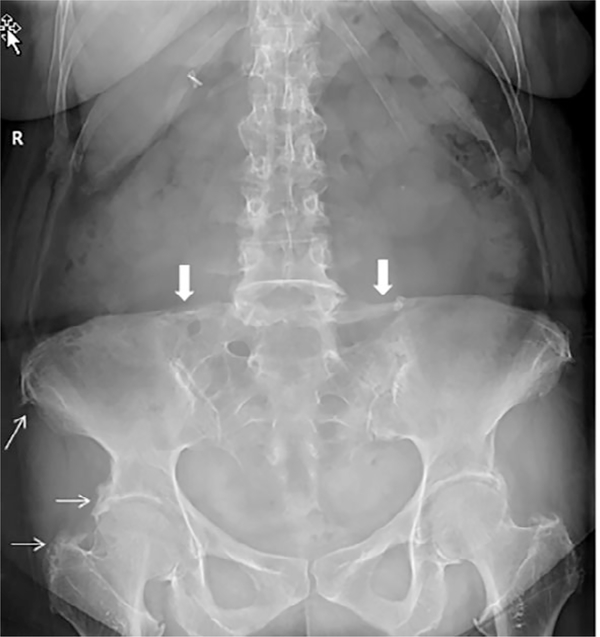
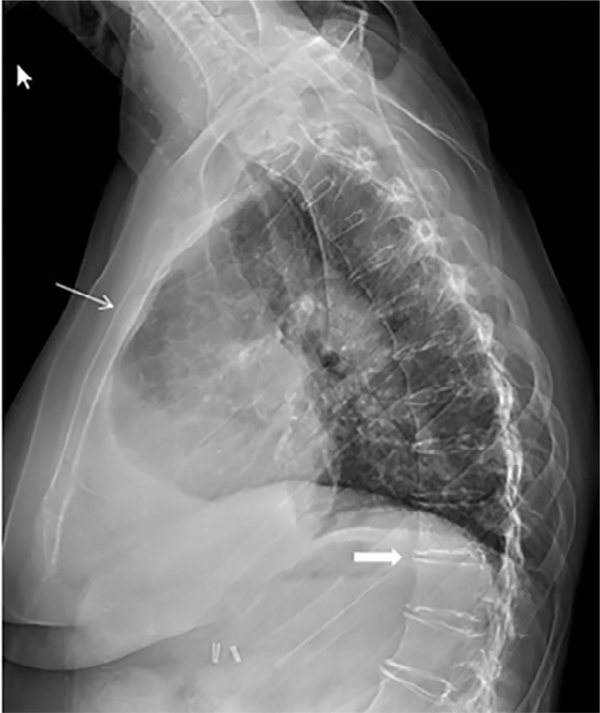
Inactivating mutations of the ENPP1 gene are associated with generalized arterial calcification of infancy (GACI) and less often autosomal-recessive hypophosphatemic rickets type 2 (ARHR2). We aimed to investigate the spectrum of phenotypes in a family with monoallelic and biallelic mutations of ENPP1 after identification through whole exome sequencing of a 54-year-old female with biallelic mutation of ENPP1, c.323G > T; p.Cys108Phe and c.1441C > T; p.Arg481Trp. Including the proband, 2 subjects had biallelic mutations, 5 had monoallelic mutations, and 2 had no mutation of ENPP1. The maternal mutation, a known pathogenic variant associated with GACI, was found in 3 subjects with monoallelic mutations, while the paternal mutation, which was not previously reported, was present in 2 subjects with monoallelic mutations. Both subjects with biallelic mutations had bowing of bilateral femurs, periarticular mineral deposition, normocalcemic primary hyperparathyroidism with multigland parathyroidectomy, increased carotid intima-media thickness, and enthesopathy was also noted in one subject. Intact FGF23 was elevated in both subjects with biallelic mutations, while C-terminal FGF23 was only elevated in one and PPi was reduced in one. Subjects with monoallelic mutations did not have periarticular calcifications or bone deformities. To conclude, patients with biallelic GACI causing mutations can survive well into adulthood, and despite the same biallelic ENPP1 pathogenic variants, clinical and biochemical manifestations can significantly differ, and include enthesopathy and primary hyperparathyroidism, which have not been previously described. Although carriers of monoallelic ENPP1 variants appear unaffected by classic disease manifestations, there may be subtle biochemical and clinical findings that warrant further investigation. © 2019 American Society for Bone and Mineral Research.
Keywords: ENPP1; GENERALIZED ARTERIAL CALCIFICATION; GENETIC; HYPOPHOSPHATEMIA; RICKETS.
© 2019 American Society for Bone and Mineral Research.
Figures
References
-
- Caswell AM, Whyte MP, Russell RG. Hypophosphatasia and the extracellular metabolism of inorganic pyrophosphate: clinical and laboratory aspects. Crit Rev Clin Lab Sci. 1991;28(3):175–232. - PubMed
-
- Ruf N, Uhlenberg B, Terkeltaub R, Nurnberg P, Rutsch F. The mutational spectrum of ENPP1 as arising after the analysis of 23 unrelated patients with generalized arterial calcification of infancy (GACI). Hum Mutat. 2005;25(1):98. - PubMed
-
- Saito T, Shimizu Y, Hori M, et al. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone. 2011;49(4): 913–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
