

Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease
- PMID: 31827005
- PMCID: PMC7011687
- DOI: 10.1212/WNL.0000000000008672
Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease
Erratum in
-
Targeted Next-Generation Sequencing Panels in the Diagnosis of Charcot-Marie-Tooth Disease.Neurology. 2022 Mar 1;98(9):384. doi: 10.1212/WNL.0000000000010635. Epub 2020 Aug 11. Neurology. 2022. PMID: 32788248 Free PMC article. No abstract available.
-
Dual Publication: Spinal Cord Injury, Vertebral Artery Dissection, and Cerebellar Strokes After Chiropractic Manipulation.Neurology. 2022 Jul 5;99(1):42. doi: 10.1212/WNL.0000000000200334. Epub 2022 Apr 4. Neurology. 2022. PMID: 35379764 Free PMC article. No abstract available.
Abstract
Objective: To investigate the effectiveness of targeted next-generation sequencing (NGS) panels in achieving a molecular diagnosis in Charcot-Marie-Tooth disease (CMT) and related disorders in a clinical setting.
Methods: We prospectively enrolled 220 patients from 2 tertiary referral centers, one in London, United Kingdom (n = 120), and one in Iowa (n = 100), in whom a targeted CMT NGS panel had been requested as a diagnostic test. PMP22 duplication/deletion was previously excluded in demyelinating cases. We reviewed the genetic and clinical data upon completion of the diagnostic process.
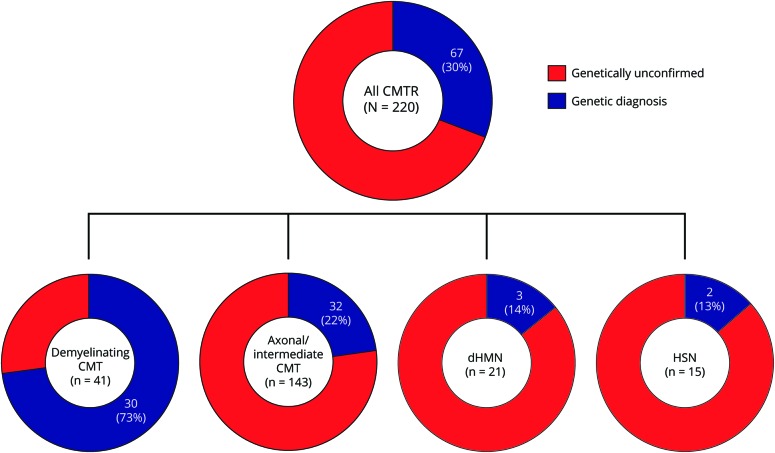
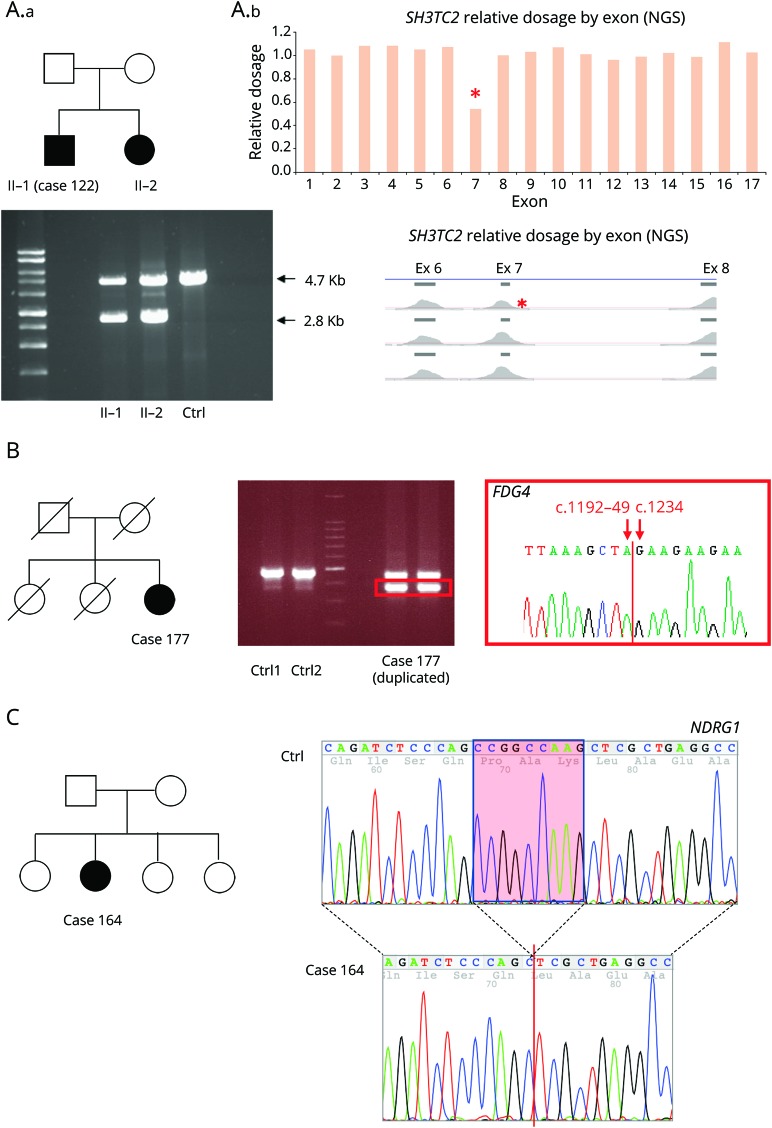
Results: After targeted NGS sequencing, a definite molecular diagnosis, defined as a pathogenic or likely pathogenic variant, was reached in 30% of cases (n = 67). The diagnostic rate was similar in London (32%) and Iowa (29%). Variants of unknown significance were found in an additional 33% of cases. Mutations in GJB1, MFN2, and MPZ accounted for 39% of cases that received genetic confirmation, while the remainder of positive cases had mutations in diverse genes, including SH3TC2, GDAP1, IGHMBP2, LRSAM1, FDG4, and GARS, and another 12 less common genes. Copy number changes in PMP22, MPZ, MFN2, SH3TC2, and FDG4 were also accurately detected. A definite genetic diagnosis was more likely in cases with an early onset, a positive family history of neuropathy or consanguinity, and a demyelinating neuropathy.
Conclusions: NGS panels are effective tools in the diagnosis of CMT, leading to genetic confirmation in one-third of cases negative for PMP22 duplication/deletion, thus highlighting how rarer and previously undiagnosed subtypes represent a relevant part of the genetic landscape of CMT.
Copyright © 2019 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the American Academy of Neurology.
Figures
References
-
- Pisciotta C, Shy ME. Neuropathy. Handb Clin Neurol 2018;148:653–665. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical