

Comprehensive detection of recurring genomic abnormalities: a targeted sequencing approach for multiple myeloma
- PMID: 31827071
- PMCID: PMC6906304
- DOI: 10.1038/s41408-019-0264-y
Comprehensive detection of recurring genomic abnormalities: a targeted sequencing approach for multiple myeloma
Erratum in
-
Correction: Comprehensive detection of recurring genomic abnormalities: a targeted sequencing approach for multiple myeloma.Blood Cancer J. 2020 Jan 30;10(1):11. doi: 10.1038/s41408-020-0279-4. Blood Cancer J. 2020. PMID: 32001687 Free PMC article.
Abstract
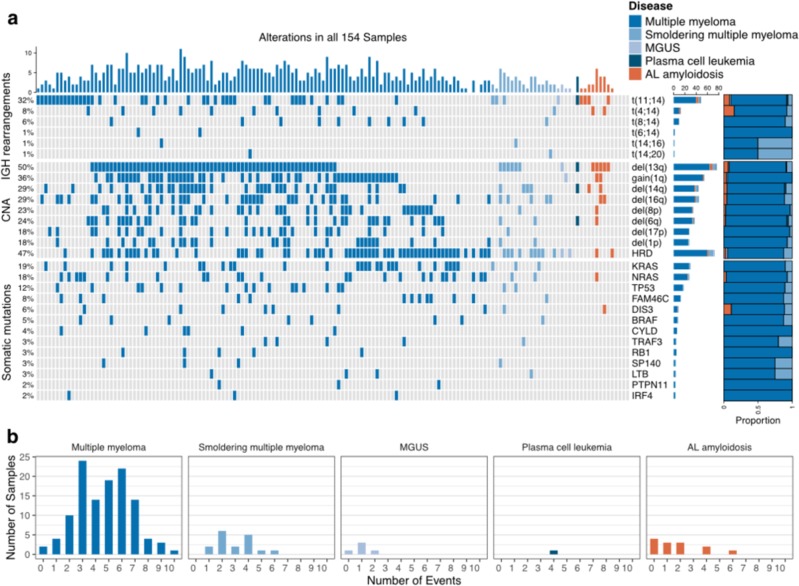
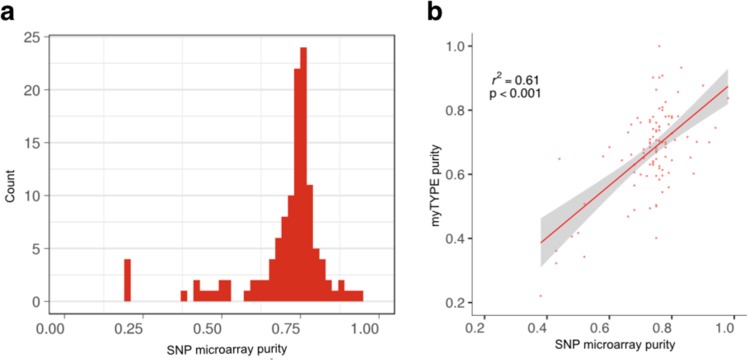
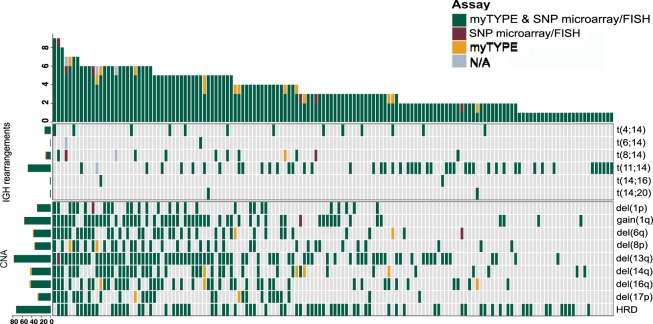
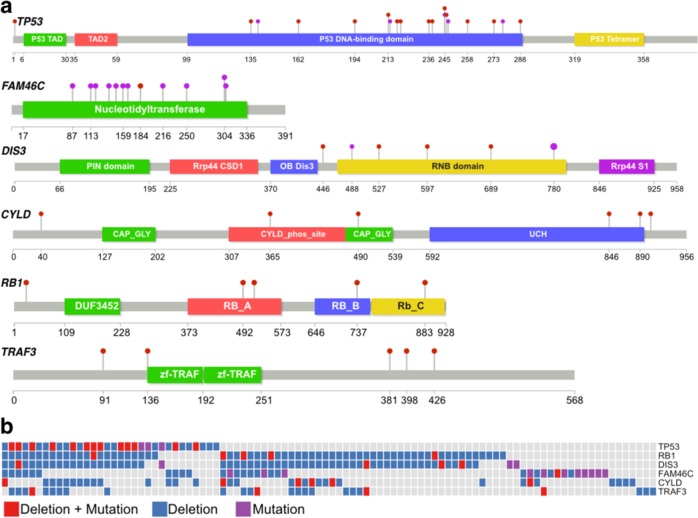
Recent genomic research efforts in multiple myeloma have revealed clinically relevant molecular subgroups beyond conventional cytogenetic classifications. Implementing these advances in clinical trial design and in routine patient care requires a new generation of molecular diagnostic tools. Here, we present a custom capture next-generation sequencing (NGS) panel designed to identify rearrangements involving the IGH locus, arm level, and focal copy number aberrations, as well as frequently mutated genes in multiple myeloma in a single assay. We sequenced 154 patients with plasma cell disorders and performed a head-to-head comparison with the results from conventional clinical assays, i.e., fluorescent in situ hybridization (FISH) and single-nucleotide polymorphism (SNP) microarray. Our custom capture NGS panel had high sensitivity (>99%) and specificity (>99%) for detection of IGH translocations and relevant chromosomal gains and losses in multiple myeloma. In addition, the assay was able to capture novel genomic markers associated with poor outcome such as bi-allelic events involving TP53. In summary, we show that a multiple myeloma designed custom capture NGS panel can detect IGH translocations and CNAs with very high concordance in relation to FISH and SNP microarrays and importantly captures the most relevant and recurrent somatic mutations in multiple myeloma rendering this approach highly suitable for clinical application in the modern era.
Conflict of interest statement
M.H. has received funding from the Multiple Myeloma Research Foundation, the Swedish Research Council, the Swedish Cancer Society, Karolinska Institute Foundations, and the Swedish Blood Cancer Foundation. N.B. has received Honoraria/ad boards from: Celgene, Janssen, Novartis. E.H. is a Josie Robertson Investigator, an EHA Fellow, and an ASH Scholar. O.L. has received research funding from: National Institutes of Health (NIH), U.S. Food and Drug Administration (FDA), Multiple Myeloma Research Foundation (MMRF), International Myeloma Foundation (IMF), Leukemia and Lymphoma Society (LLS), Perelman Family Foundation, Rising Tides Foundation, Amgen, Celgene, Janssen, Takeda, Glenmark, Seattle Genetics, Karyopharm; Honoraria/ad boards: Adaptive, Amgen, Binding Site, BMS, Celgene, Cellectis, Glenmark, Janssen, Juno, Pfizer; and serves on Independent Data Monitoring Committees (IDMCs) for clinical trials lead by Takeda, Merck, Janssen, and Theradex. There is a pending patent application regarding the myTYPE assay (M.H., E.P., and O.L.). The remaining authors declare no relevant conflicts of interest.
Figures

References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
