

DNA methylation directs microRNA biogenesis in mammalian cells
- PMID: 31827083
- PMCID: PMC6906426
- DOI: 10.1038/s41467-019-13527-1
DNA methylation directs microRNA biogenesis in mammalian cells
Abstract
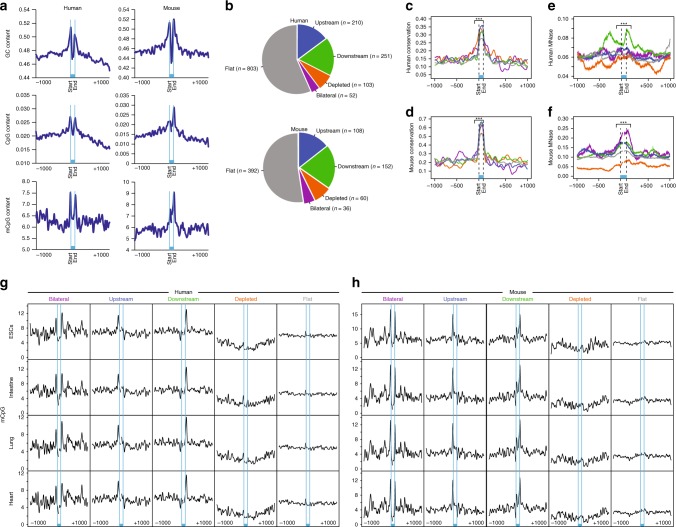
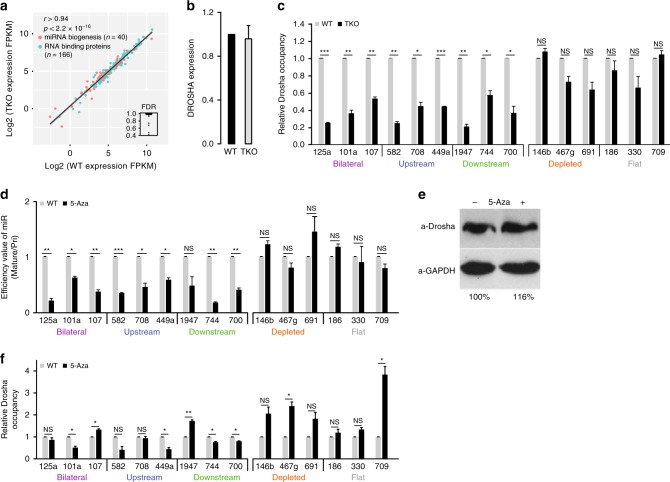
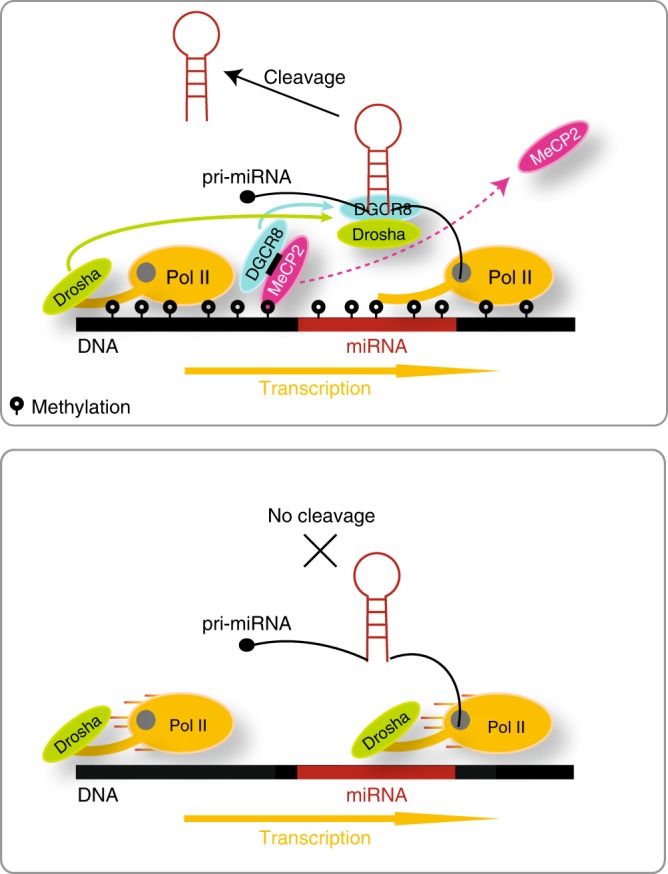
MicroRNA (miRNA) biogenesis initiates co-transcriptionally, but how the Microprocessor machinery pinpoints the locations of short precursor miRNA sequences within long flanking regions of the transcript is not known. Here we show that miRNA biogenesis depends on DNA methylation. When the regions flanking the miRNA coding sequence are highly methylated, the miRNAs are more highly expressed, have greater sequence conservation, and are more likely to drive cancer-related phenotypes than miRNAs encoded by unmethylated loci. We show that the removal of DNA methylation from miRNA loci leads to their downregulation. Further, we found that MeCP2 binding to methylated miRNA loci halts RNA polymerase II elongation, leading to enhanced processing of the primary miRNA by Drosha. Taken together, our data reveal that DNA methylation directly affects miRNA biogenesis.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Voorhoeve PM, Agami R. Classifying microRNAs in cancer: the good, the bad and the ugly. Biochim. Biophys. Acta. 2007;1775:274–282. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
