

Improving the odds of drug development success through human genomics: modelling study
- PMID: 31827124
- PMCID: PMC6906499
- DOI: 10.1038/s41598-019-54849-w
Improving the odds of drug development success through human genomics: modelling study
Abstract
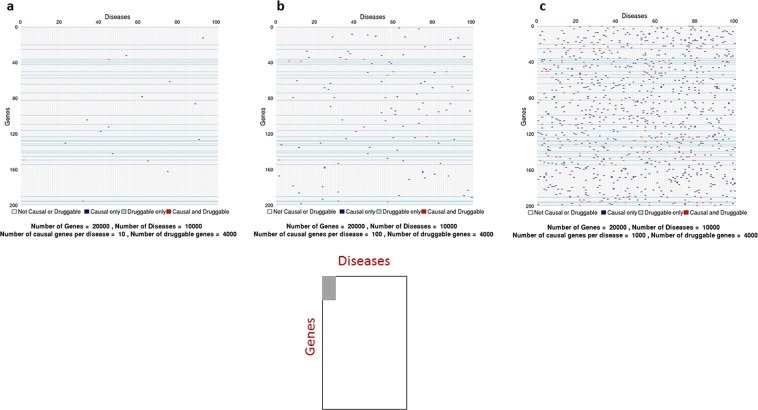
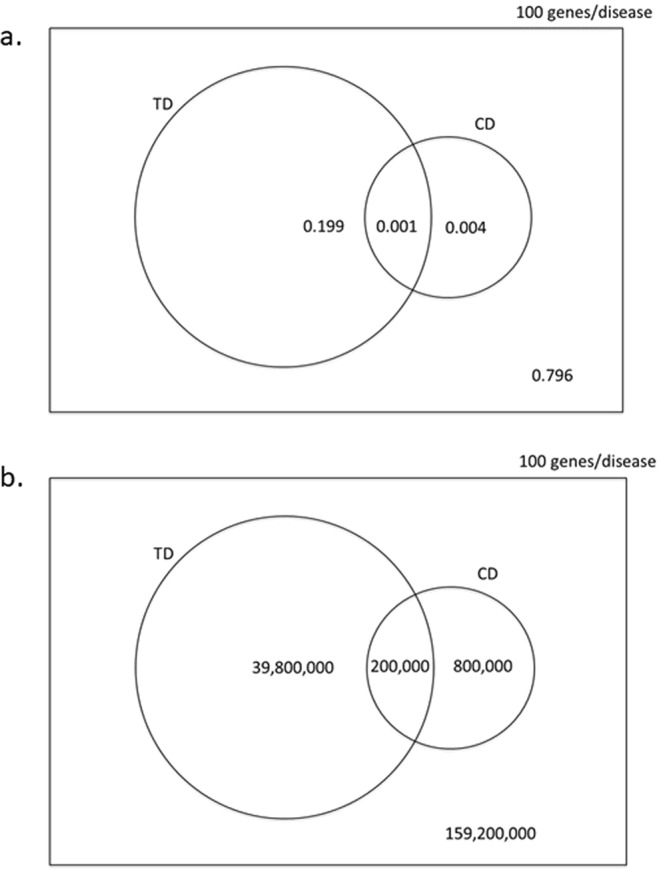
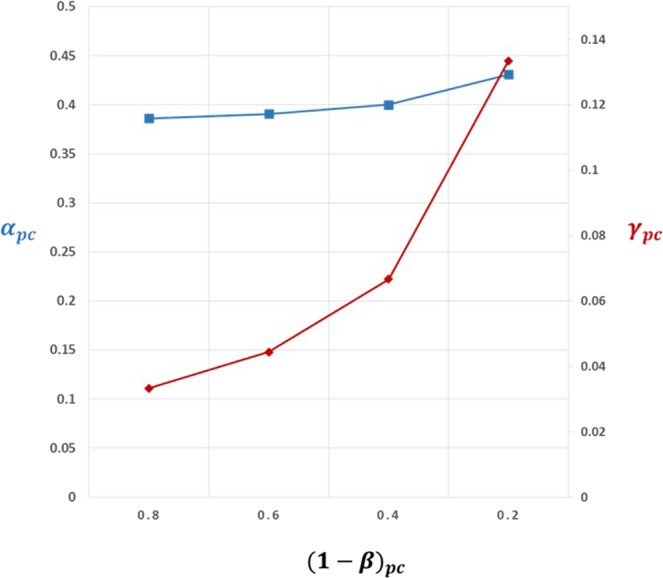
Lack of efficacy in the intended disease indication is the major cause of clinical phase drug development failure. Explanations could include the poor external validity of pre-clinical (cell, tissue, and animal) models of human disease and the high false discovery rate (FDR) in preclinical science. FDR is related to the proportion of true relationships available for discovery (γ), and the type 1 (false-positive) and type 2 (false negative) error rates of the experiments designed to uncover them. We estimated the FDR in preclinical science, its effect on drug development success rates, and improvements expected from use of human genomics rather than preclinical studies as the primary source of evidence for drug target identification. Calculations were based on a sample space defined by all human diseases - the 'disease-ome' - represented as columns; and all protein coding genes - 'the protein-coding genome'- represented as rows, producing a matrix of unique gene- (or protein-) disease pairings. We parameterised the space based on 10,000 diseases, 20,000 protein-coding genes, 100 causal genes per disease and 4000 genes encoding druggable targets, examining the effect of varying the parameters and a range of underlying assumptions, on the inferences drawn. We estimated γ, defined mathematical relationships between preclinical FDR and drug development success rates, and estimated improvements in success rates based on human genomics (rather than orthodox preclinical studies). Around one in every 200 protein-disease pairings was estimated to be causal (γ = 0.005) giving an FDR in preclinical research of 92.6%, which likely makes a major contribution to the reported drug development failure rate of 96%. Observed success rate was only slightly greater than expected for a random pick from the sample space. Values for γ back-calculated from reported preclinical and clinical drug development success rates were also close to the a priori estimates. Substituting genome wide (or druggable genome wide) association studies for preclinical studies as the major information source for drug target identification was estimated to reverse the probability of late stage failure because of the more stringent type 1 error rate employed and the ability to interrogate every potential druggable target in the same experiment. Genetic studies conducted at much larger scale, with greater resolution of disease end-points, e.g. by connecting genomics and electronic health record data within healthcare systems has the potential to produce radical improvement in drug development success rate.
Conflict of interest statement
Benevolent AI provided financial support in the form of salaries for two authors – Dr. Felix Kruger and Professor John Overington during part of the period covered by this work. Benevolent AI did not play a role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Figures

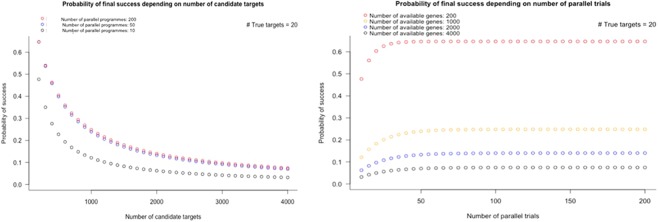
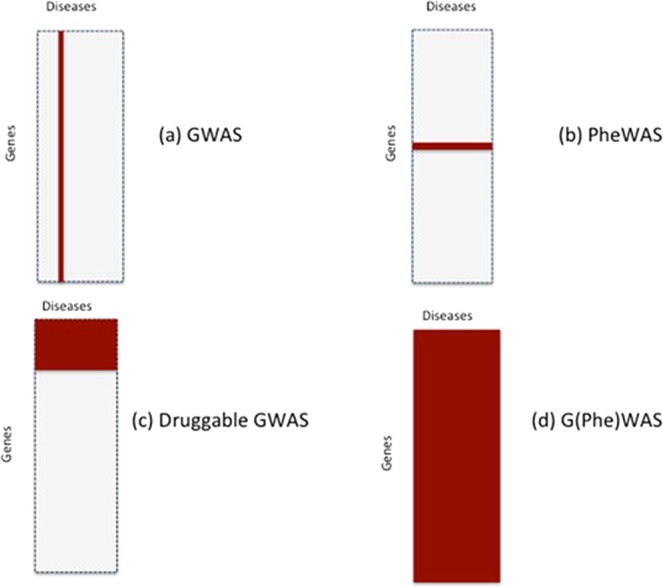
References
-
- Paul Steven M., Mytelka Daniel S., Dunwiddie Christopher T., Persinger Charles C., Munos Bernard H., Lindborg Stacy R., Schacht Aaron L. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nature Reviews Drug Discovery. 2010;9(3):203–214. doi: 10.1038/nrd3078. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
