

Multi-omics profiling of mouse gastrulation at single-cell resolution
- PMID: 31827285
- PMCID: PMC6924995
- DOI: 10.1038/s41586-019-1825-8
Multi-omics profiling of mouse gastrulation at single-cell resolution
Abstract
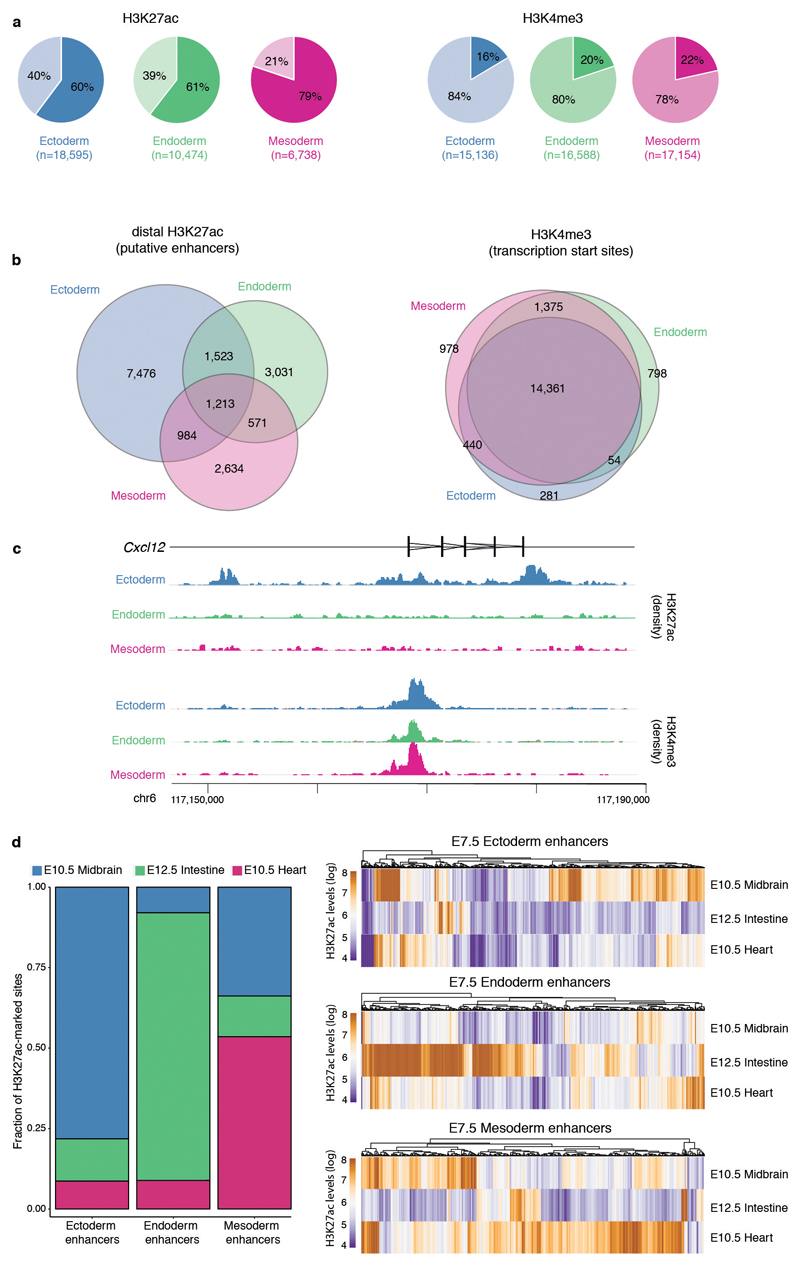
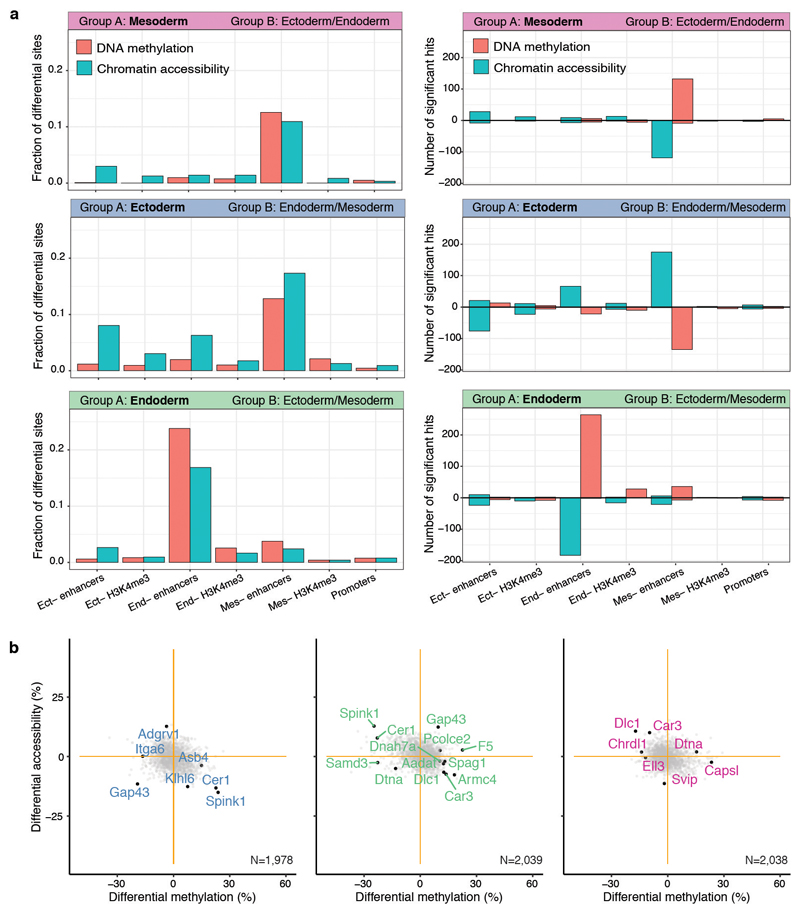
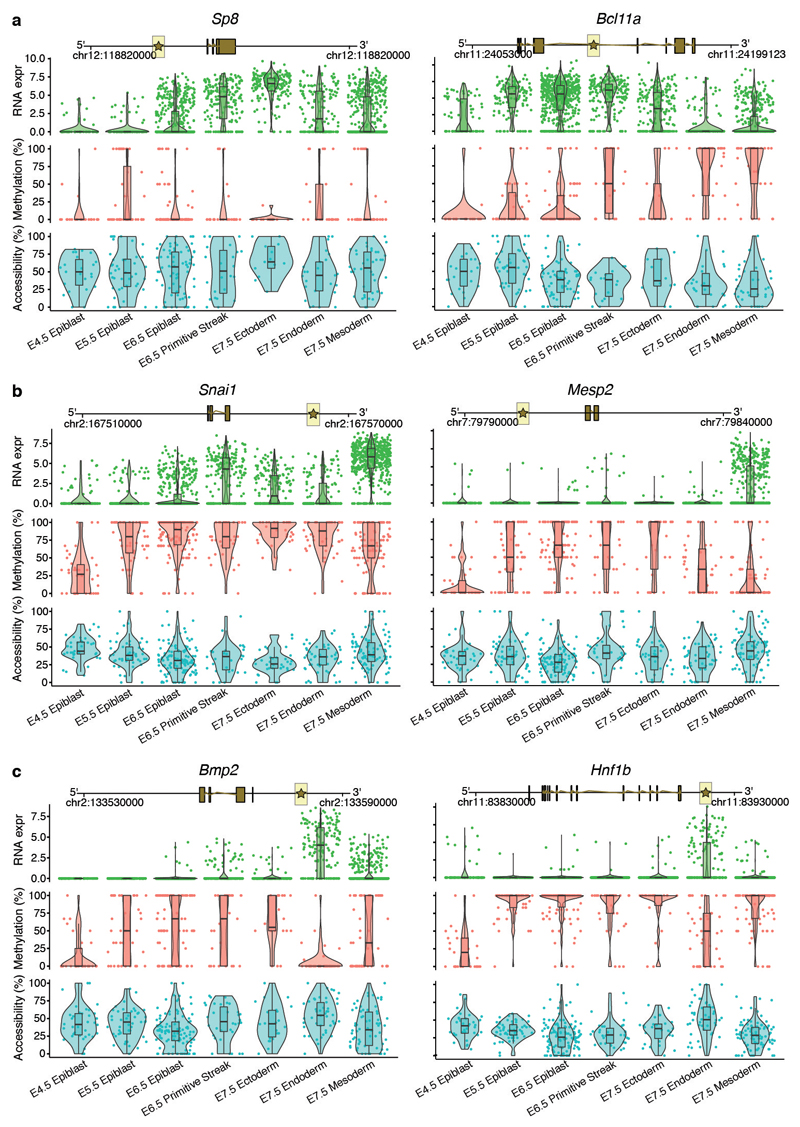
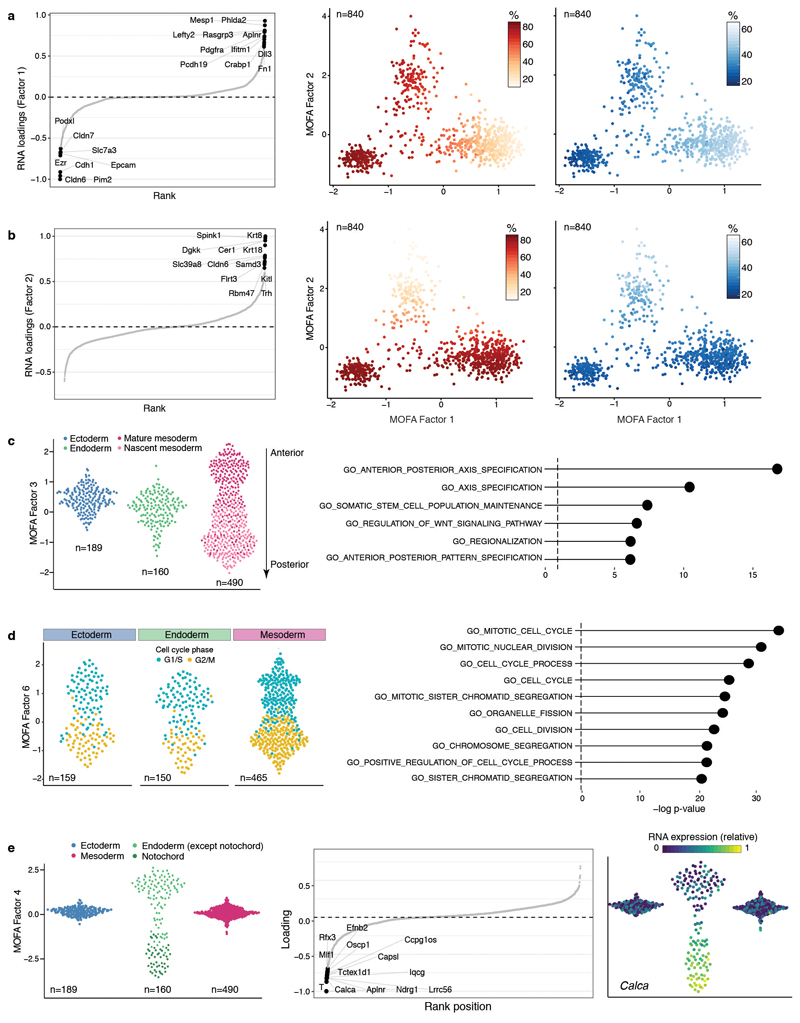
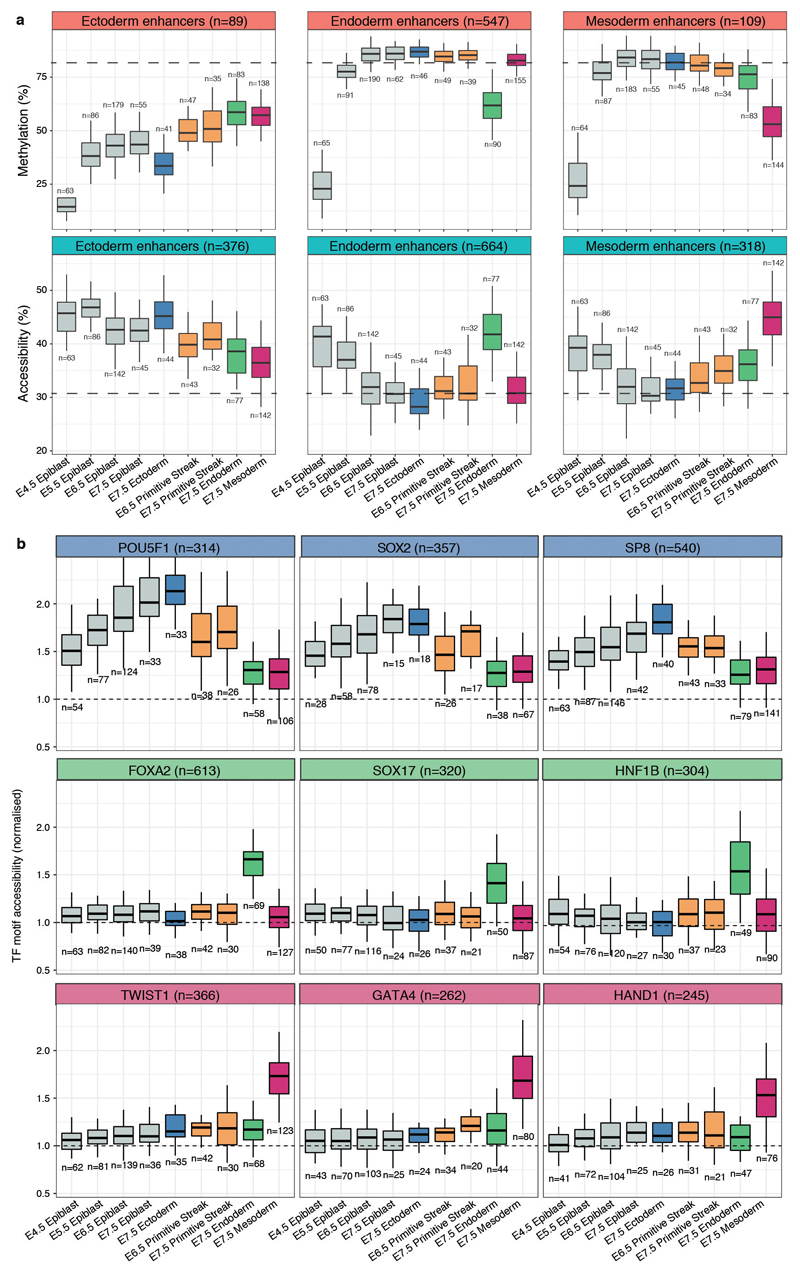
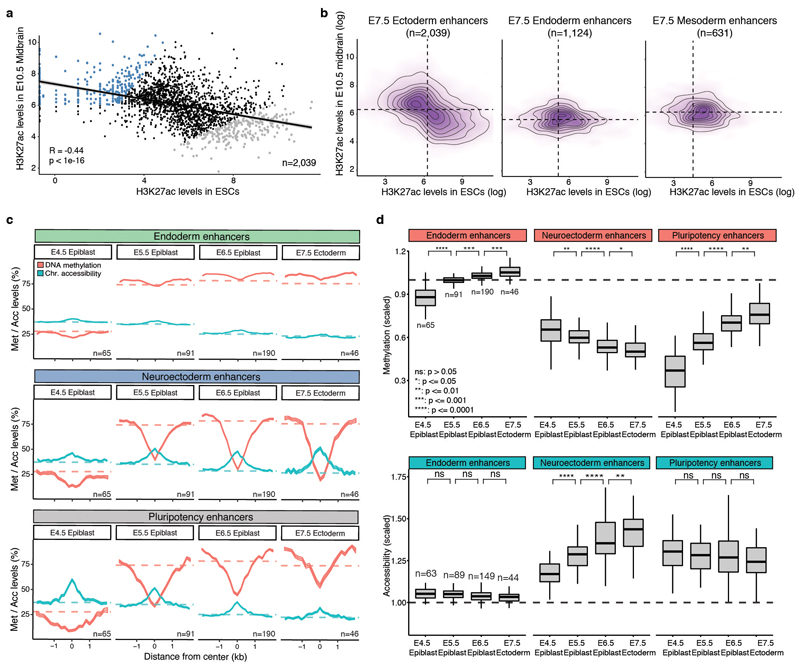
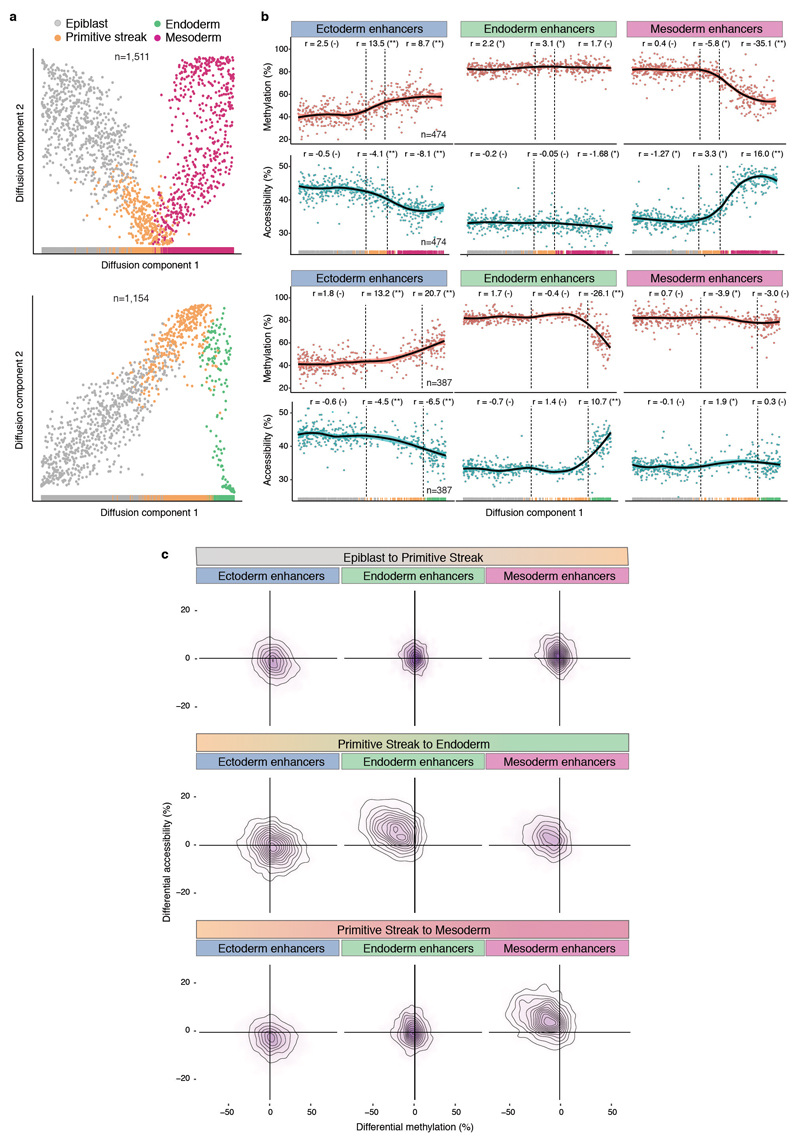
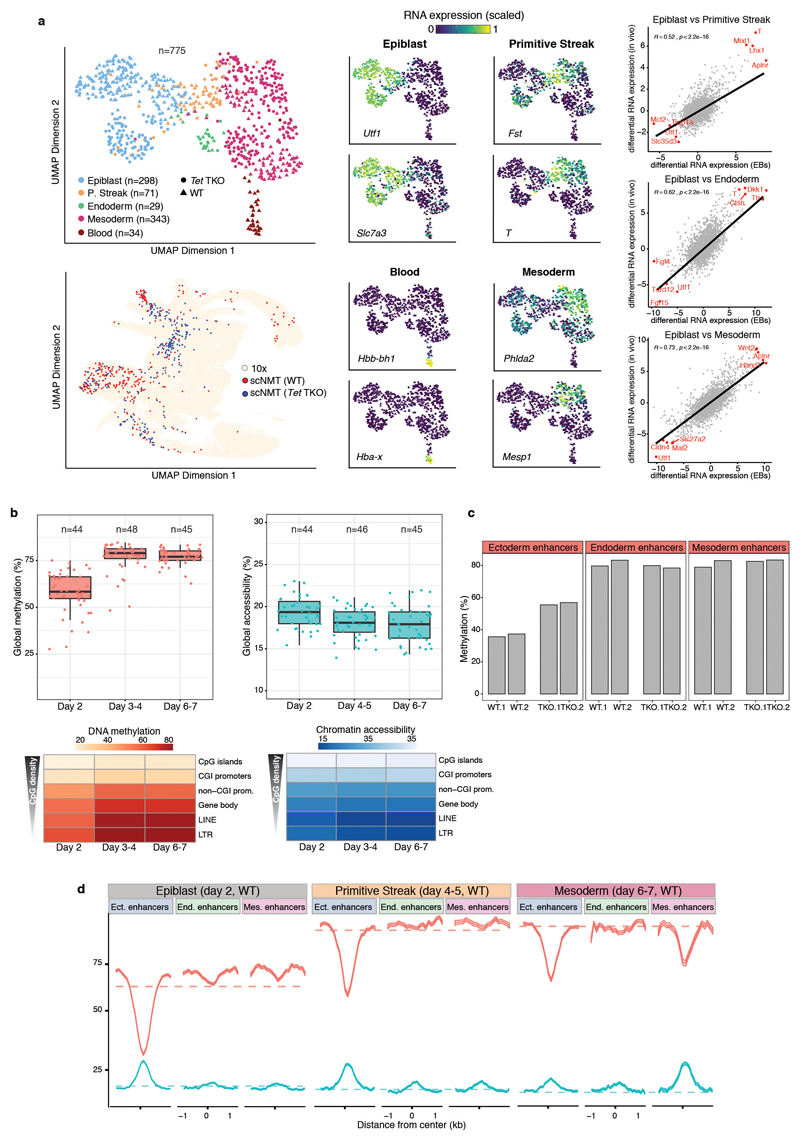
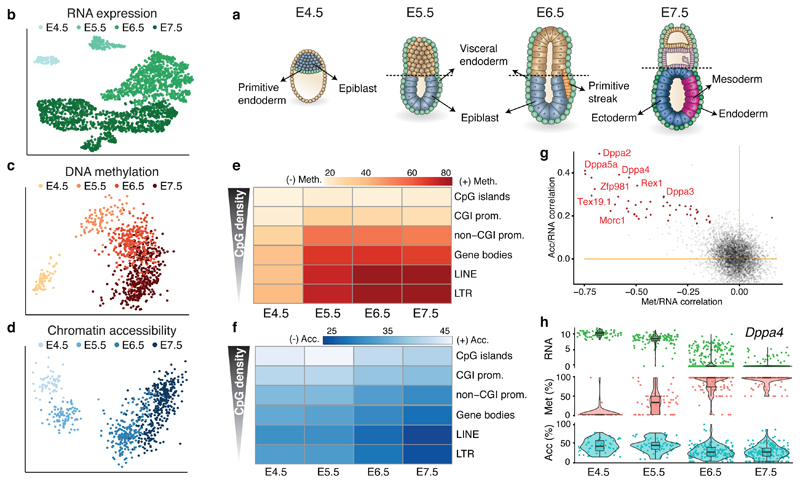
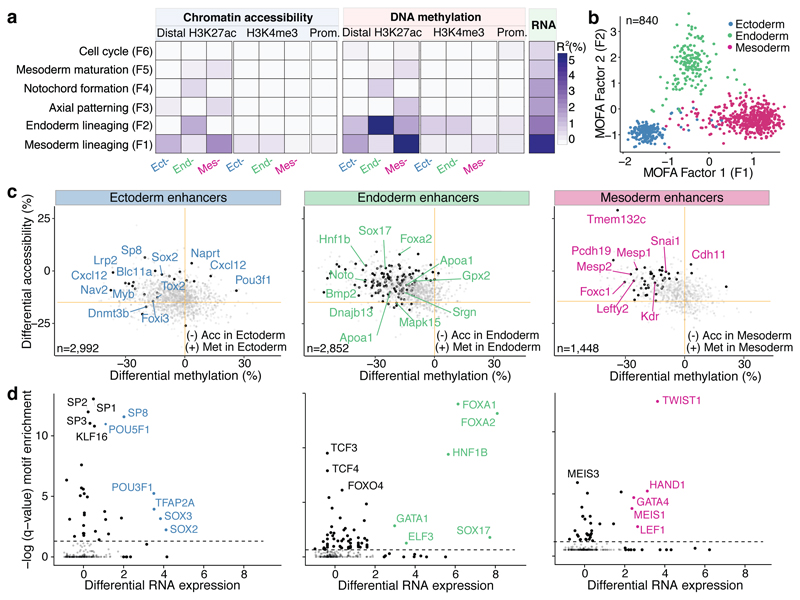
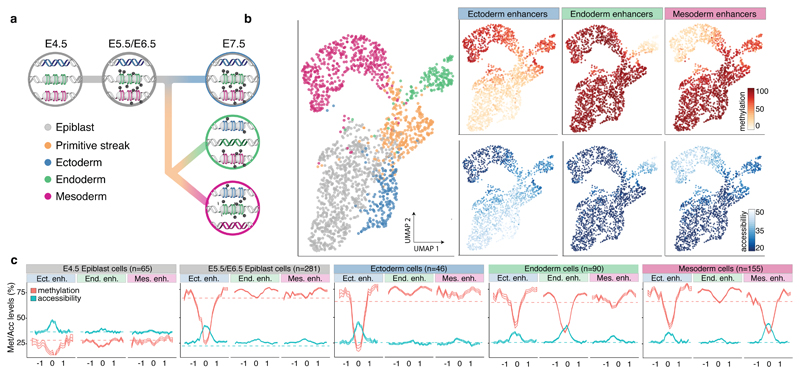
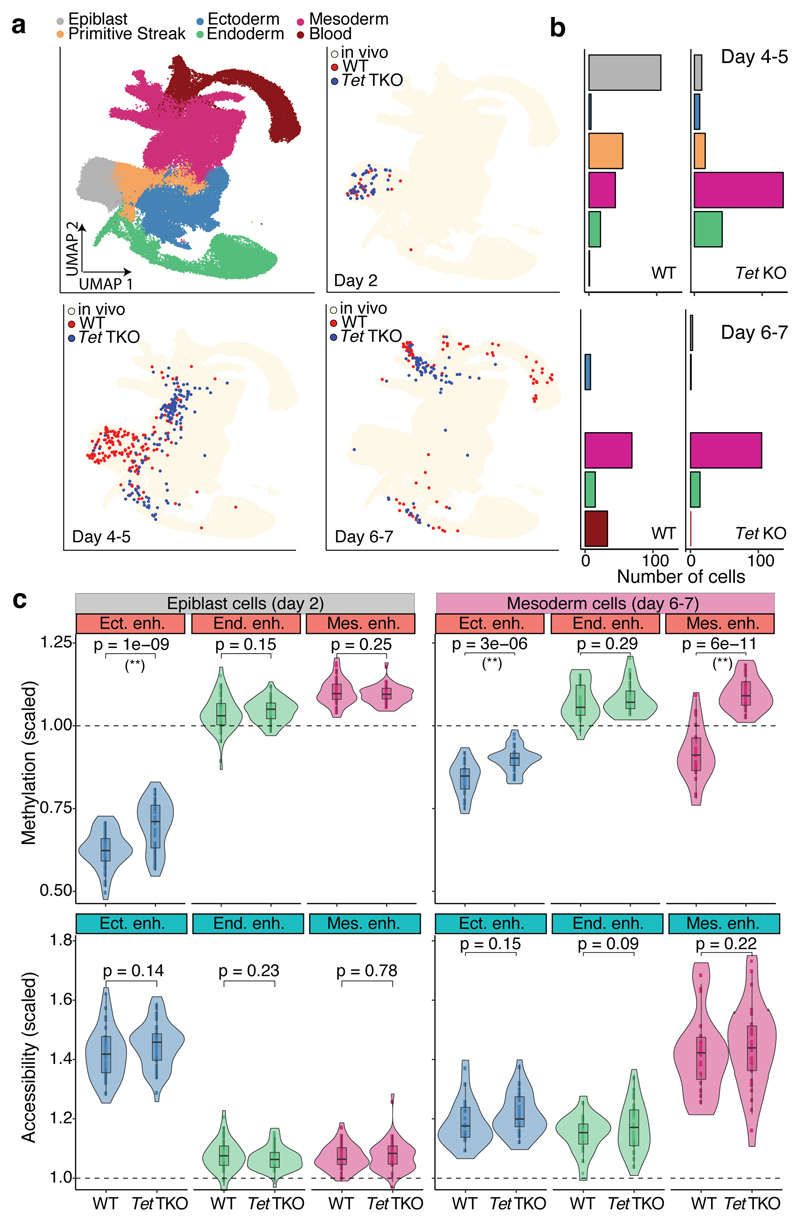
Formation of the three primary germ layers during gastrulation is an essential step in the establishment of the vertebrate body plan and is associated with major transcriptional changes1-5. Global epigenetic reprogramming accompanies these changes6-8, but the role of the epigenome in regulating early cell-fate choice remains unresolved, and the coordination between different molecular layers is unclear. Here we describe a single-cell multi-omics map of chromatin accessibility, DNA methylation and RNA expression during the onset of gastrulation in mouse embryos. The initial exit from pluripotency coincides with the establishment of a global repressive epigenetic landscape, followed by the emergence of lineage-specific epigenetic patterns during gastrulation. Notably, cells committed to mesoderm and endoderm undergo widespread coordinated epigenetic rearrangements at enhancer marks, driven by ten-eleven translocation (TET)-mediated demethylation and a concomitant increase of accessibility. By contrast, the methylation and accessibility landscape of ectodermal cells is already established in the early epiblast. Hence, regulatory elements associated with each germ layer are either epigenetically primed or remodelled before cell-fate decisions, providing the molecular framework for a hierarchical emergence of the primary germ layers.
Conflict of interest statement
W.R. is a consultant and shareholder of Cambridge Epigenetix. The remaining authors declare no competing financial interests
Figures
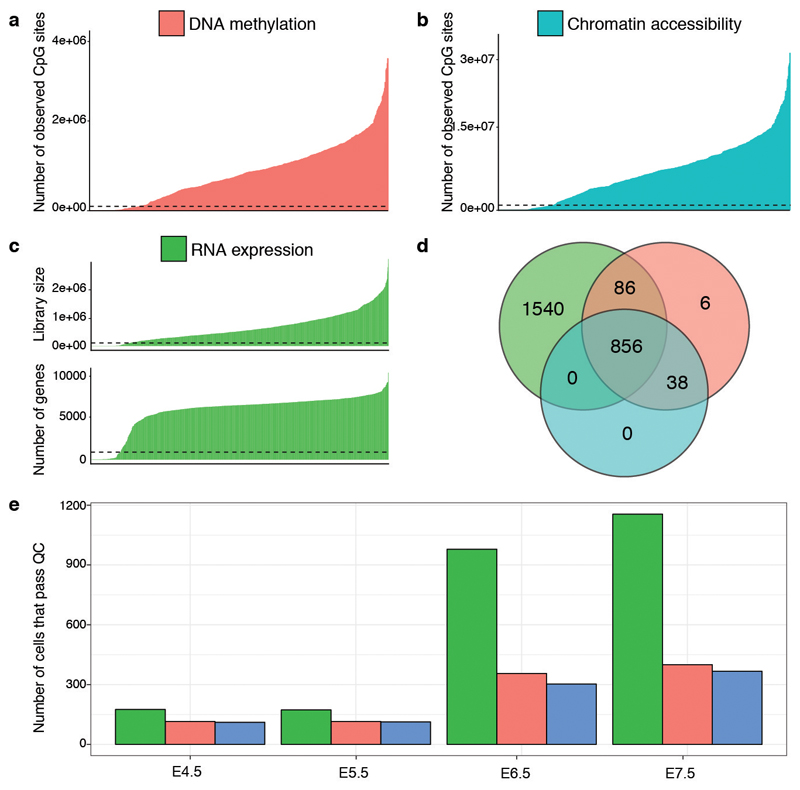
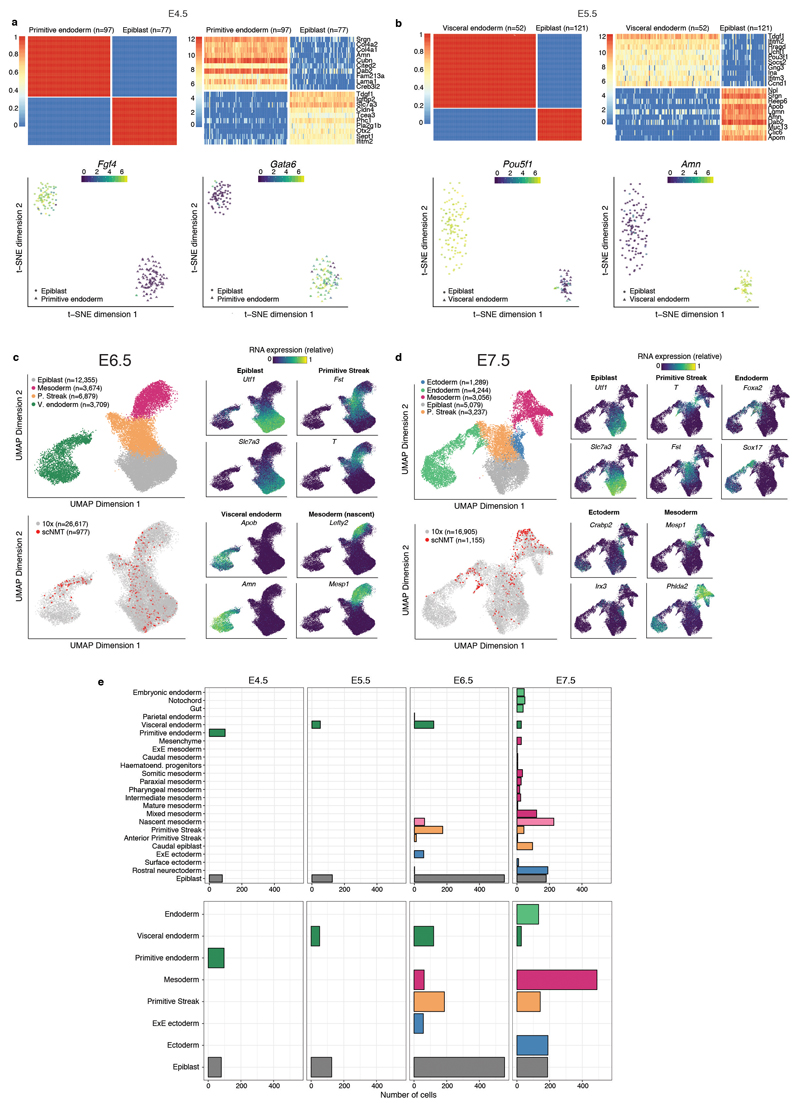
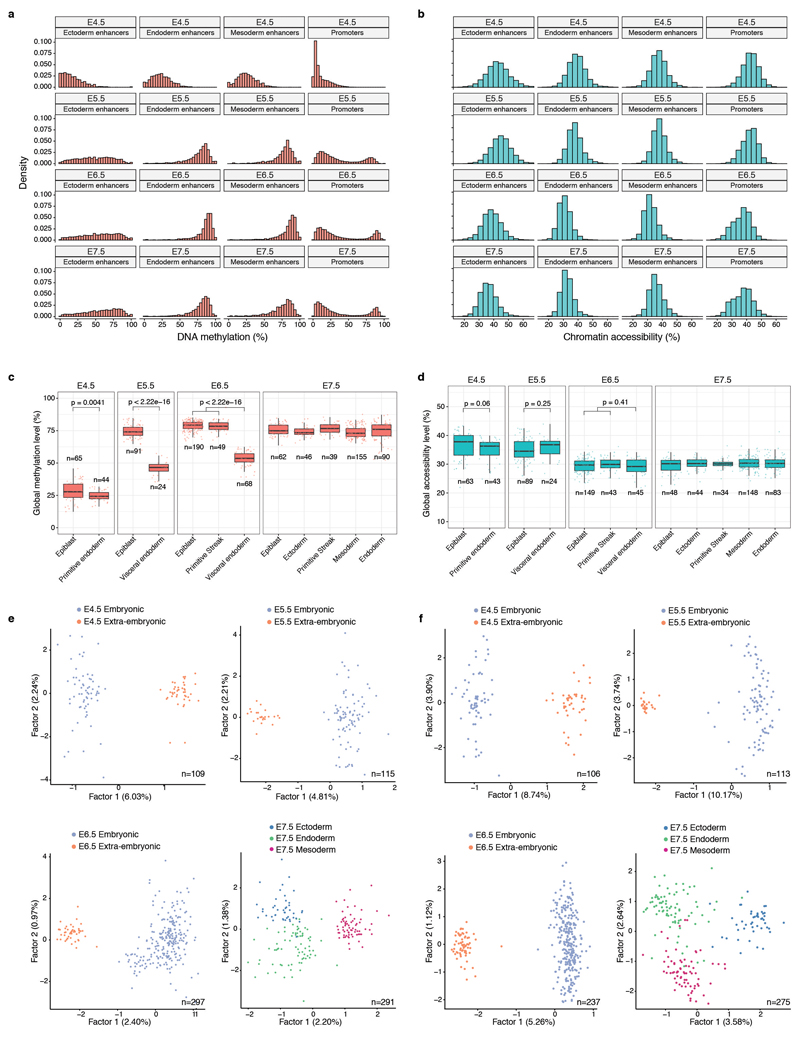
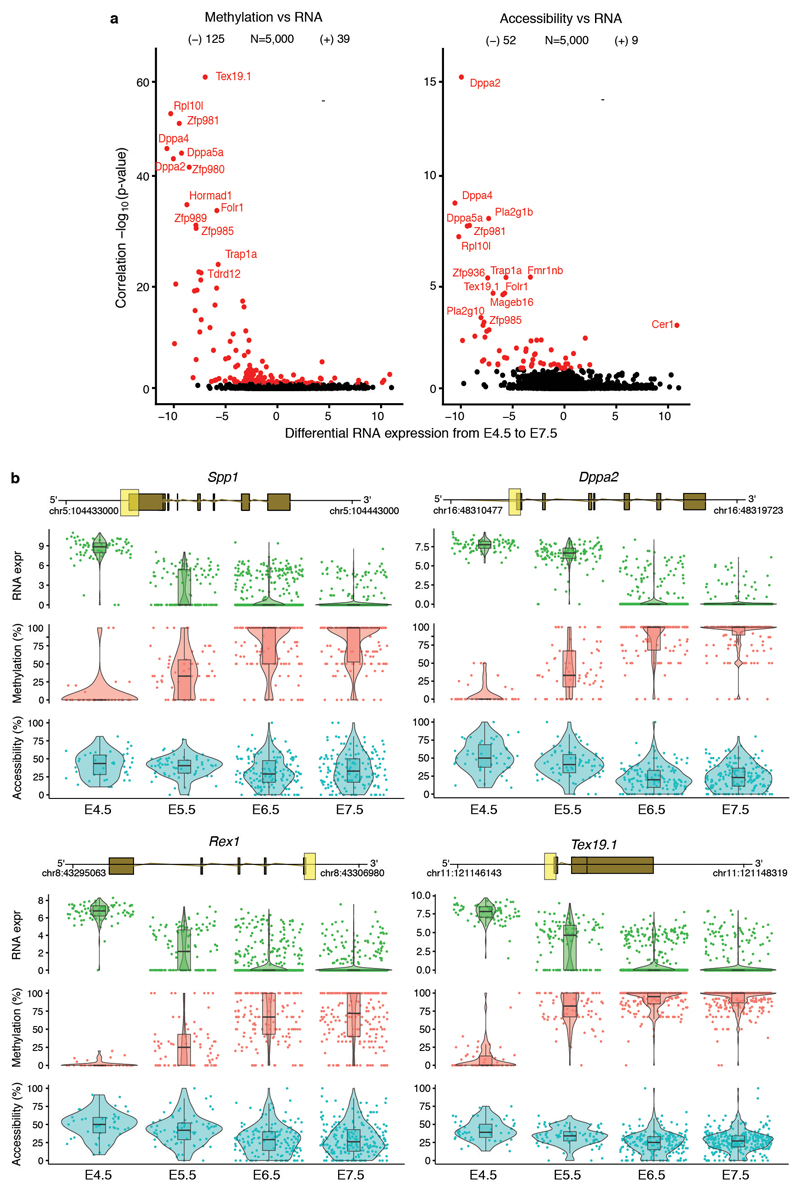
Comment in
-
Multi-omics shows the (default) way.Nat Rev Genet. 2020 Mar;21(3):134-135. doi: 10.1038/s41576-020-0211-6. Nat Rev Genet. 2020. PMID: 31925408 No abstract available.
References
-
- Peng G, et al. Spatial Transcriptome for the Molecular Annotation of Lineage Fates and Cell Identity in Mid-gastrula Mouse Embryo. Dev Cell. 2016;36:681–697. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- 22231/CRUK_/Cancer Research UK/United Kingdom
- MC_UU_00009/2/MRC_/Medical Research Council/United Kingdom
- 105031/WT_/Wellcome Trust/United Kingdom
- MC_UU_00009/1/MRC_/Medical Research Council/United Kingdom
- MR/K011332/1/MRC_/Medical Research Council/United Kingdom
- 810296/ERC_/European Research Council/International
- 108438/E/15/Z/WT_/Wellcome Trust/United Kingdom
- 210754/Z/18/Z/WT_/Wellcome Trust/United Kingdom
- MC_PC_12009/MRC_/Medical Research Council/United Kingdom
- 095645/WT_/Wellcome Trust/United Kingdom
- MR/M01536X/1/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
