

Tensin1 expression and function in chronic obstructive pulmonary disease
- PMID: 31831813
- PMCID: PMC6908681
- DOI: 10.1038/s41598-019-55405-2
Tensin1 expression and function in chronic obstructive pulmonary disease
Abstract
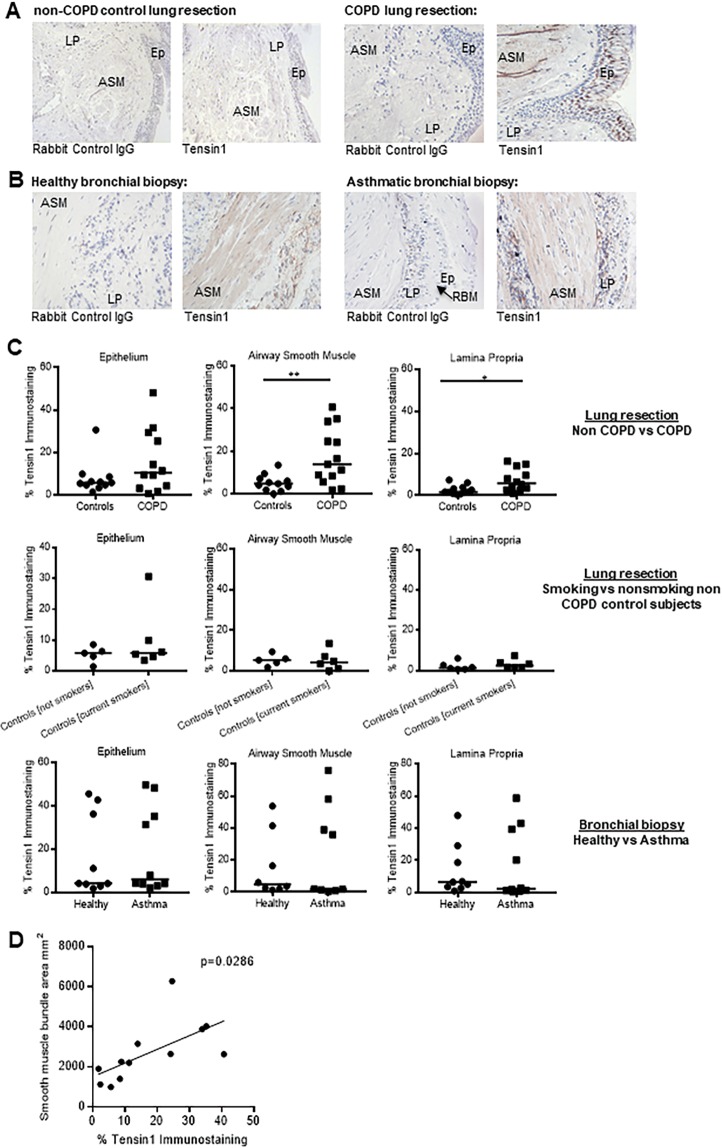
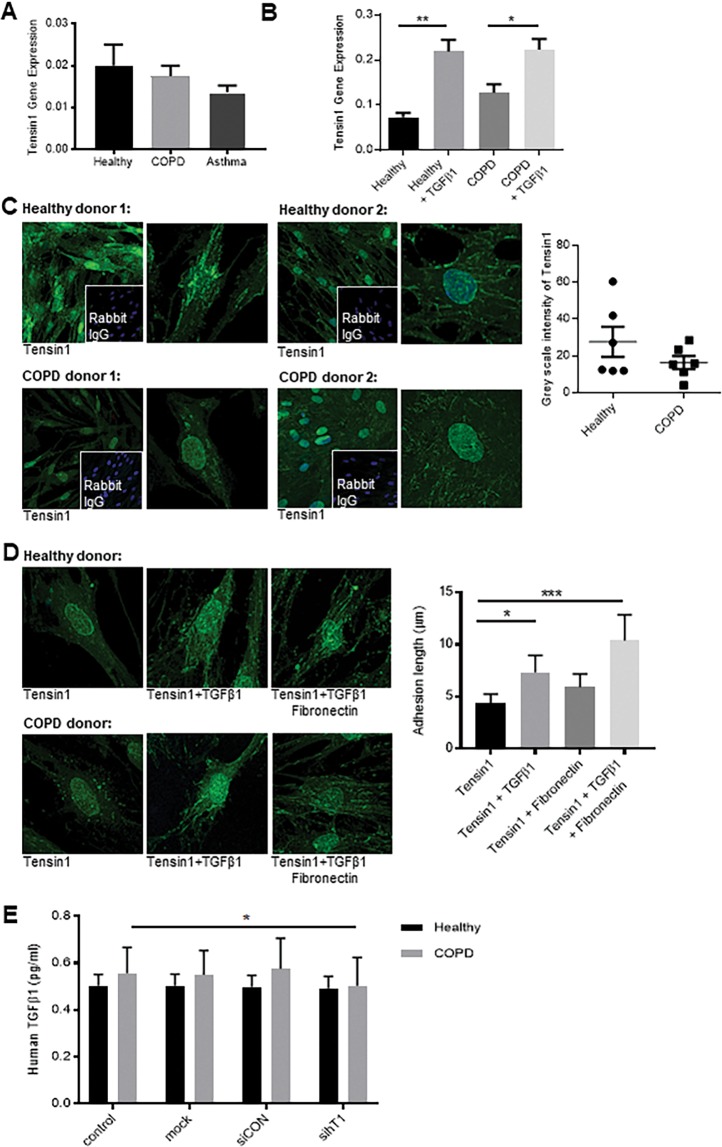
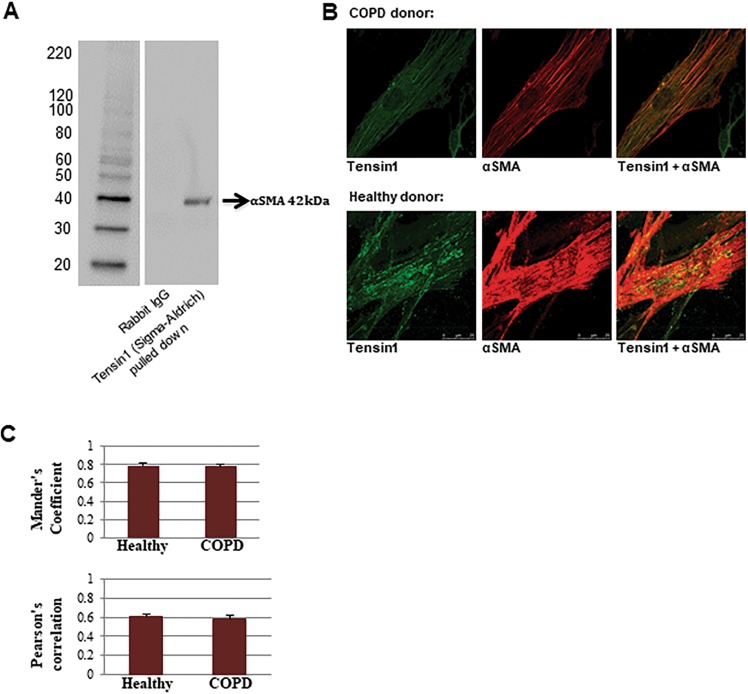
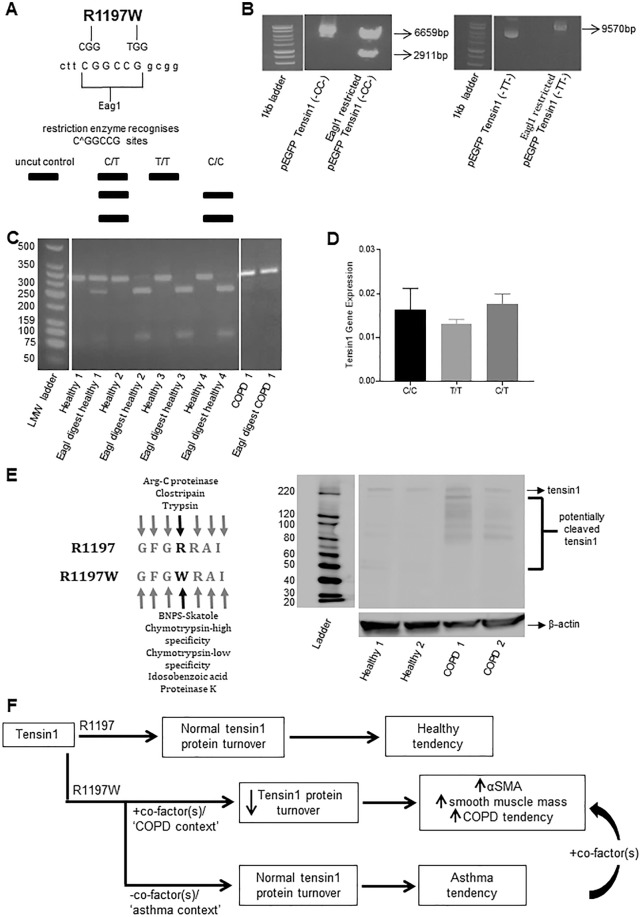
Chronic obstructive pulmonary disease (COPD) constitutes a major cause of morbidity and mortality. Genome wide association studies have shown significant associations between airflow obstruction or COPD with a non-synonymous SNP in the TNS1 gene, which encodes tensin1. However, the expression, cellular distribution and function of tensin1 in human airway tissue and cells are unknown. We therefore examined these characteristics in tissue and cells from controls and people with COPD or asthma. Airway tissue was immunostained for tensin1. Tensin1 expression in cultured human airway smooth muscle cells (HASMCs) was evaluated using qRT-PCR, western blotting and immunofluorescent staining. siRNAs were used to downregulate tensin1 expression. Tensin1 expression was increased in the airway smooth muscle and lamina propria in COPD tissue, but not asthma, when compared to controls. Tensin1 was expressed in HASMCs and upregulated by TGFβ1. TGFβ1 and fibronectin increased the localisation of tensin1 to fibrillar adhesions. Tensin1 and α-smooth muscle actin (αSMA) were strongly co-localised, and tensin1 depletion in HASMCs attenuated both αSMA expression and contraction of collagen gels. In summary, tensin1 expression is increased in COPD airways, and may promote airway obstruction by enhancing the expression of contractile proteins and their localisation to stress fibres in HASMCs.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
-
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR, Hurd SS. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med. 2001;163:1256–1276. doi: 10.1164/ajrccm.163.5.2101039. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
