

Genome wide analysis reveals heparan sulfate epimerase modulates TDP-43 proteinopathy
- PMID: 31834878
- PMCID: PMC6934317
- DOI: 10.1371/journal.pgen.1008526
Genome wide analysis reveals heparan sulfate epimerase modulates TDP-43 proteinopathy
Abstract
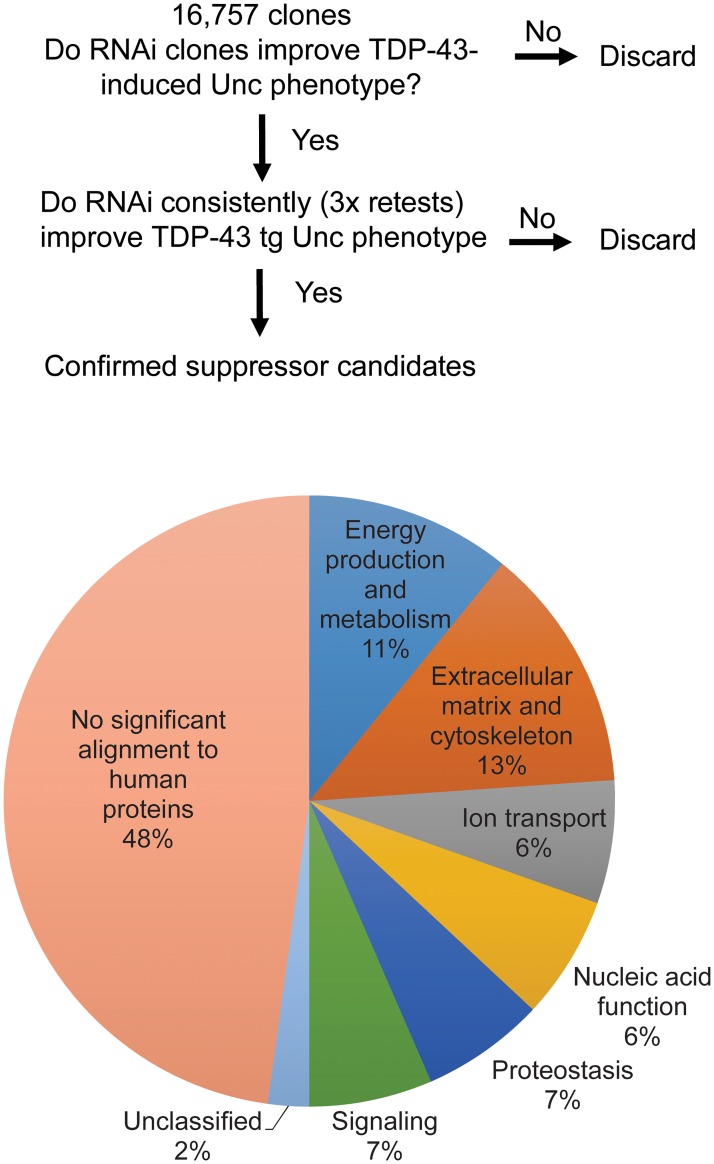
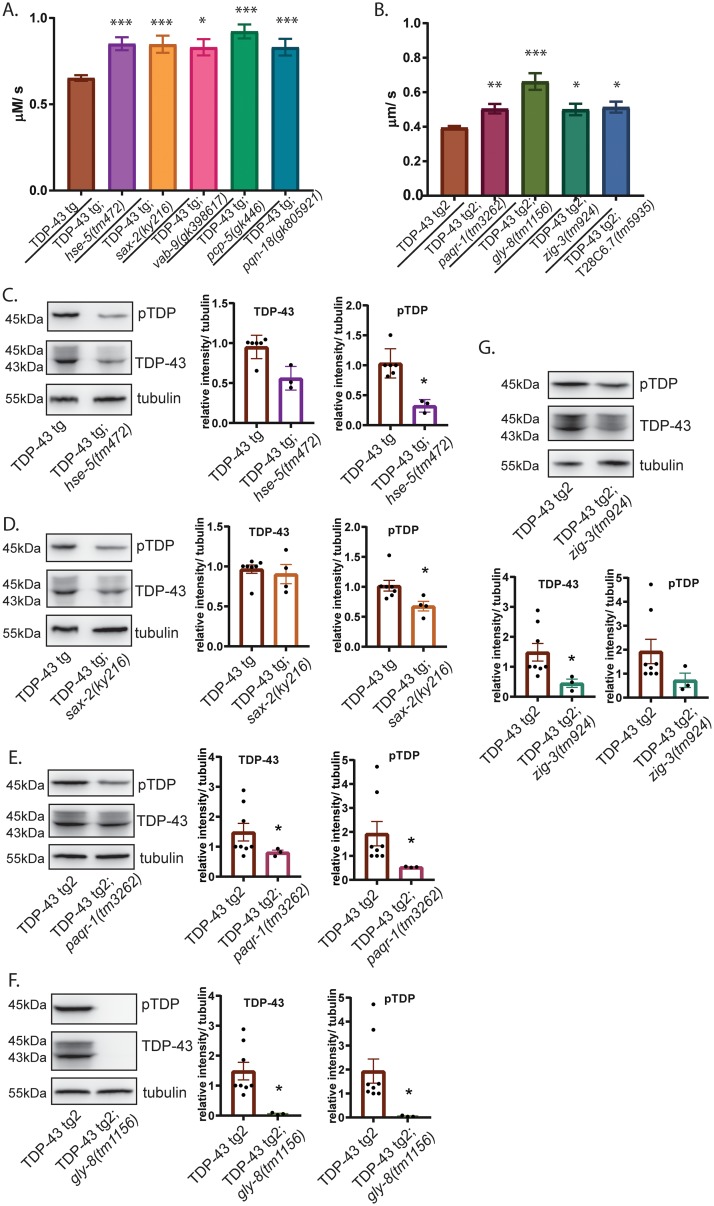
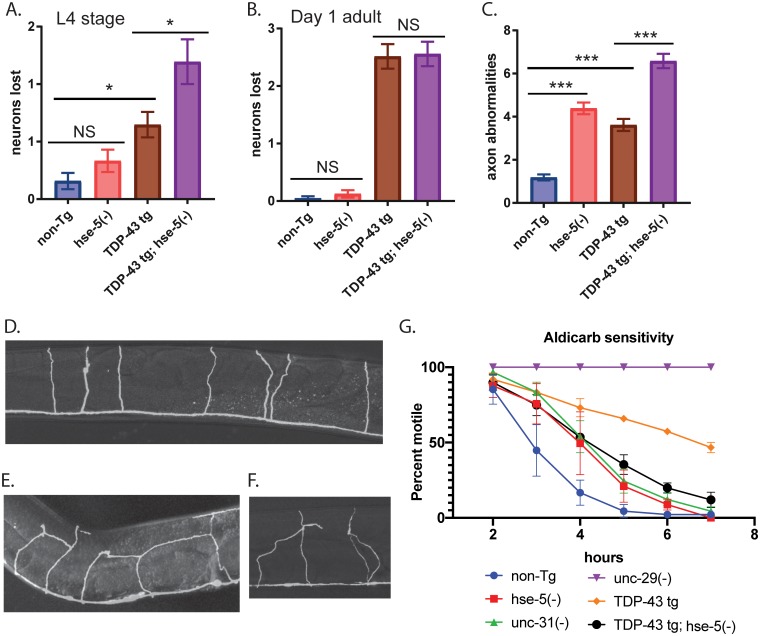
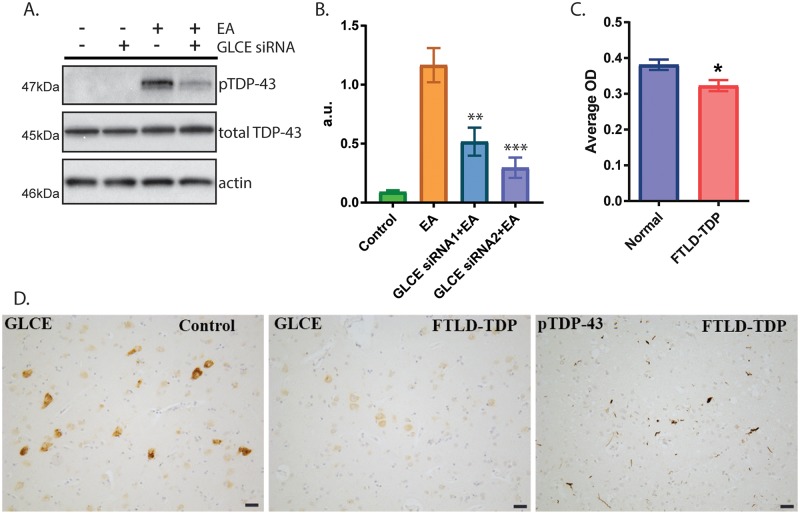
Pathological phosphorylated TDP-43 protein (pTDP) deposition drives neurodegeneration in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD-TDP). However, the cellular and genetic mechanisms at work in pathological TDP-43 toxicity are not fully elucidated. To identify genetic modifiers of TDP-43 neurotoxicity, we utilized a Caenorhabditis elegans model of TDP-43 proteinopathy expressing human mutant TDP-43 pan-neuronally (TDP-43 tg). In TDP-43 tg C. elegans, we conducted a genome-wide RNAi screen covering 16,767 C. elegans genes for loss of function genetic suppressors of TDP-43-driven motor dysfunction. We identified 46 candidate genes that when knocked down partially ameliorate TDP-43 related phenotypes; 24 of these candidate genes have conserved homologs in the human genome. To rigorously validate the RNAi findings, we crossed the TDP-43 transgene into the background of homozygous strong genetic loss of function mutations. We have confirmed 9 of the 24 candidate genes significantly modulate TDP-43 transgenic phenotypes. Among the validated genes we focused on, one of the most consistent genetic modifier genes protecting against pTDP accumulation and motor deficits was the heparan sulfate-modifying enzyme hse-5, the C. elegans homolog of glucuronic acid epimerase (GLCE). We found that knockdown of human GLCE in cultured human cells protects against oxidative stress induced pTDP accumulation. Furthermore, expression of glucuronic acid epimerase is significantly decreased in the brains of FTLD-TDP cases relative to normal controls, demonstrating the potential disease relevance of the candidate genes identified. Taken together these findings nominate glucuronic acid epimerase as a novel candidate therapeutic target for TDP-43 proteinopathies including ALS and FTLD-TDP.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
