

Computational modeling demonstrates that glioblastoma cells can survive spatial environmental challenges through exploratory adaptation
- PMID: 31836713
- PMCID: PMC6911112
- DOI: 10.1038/s41467-019-13726-w
Computational modeling demonstrates that glioblastoma cells can survive spatial environmental challenges through exploratory adaptation
Abstract
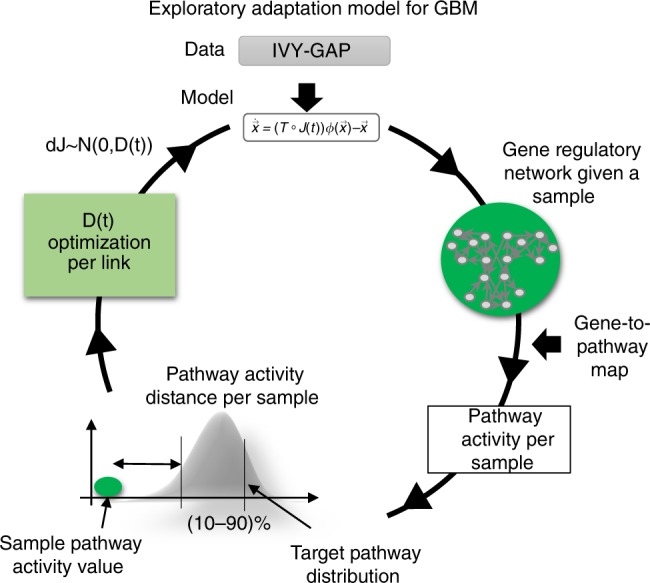
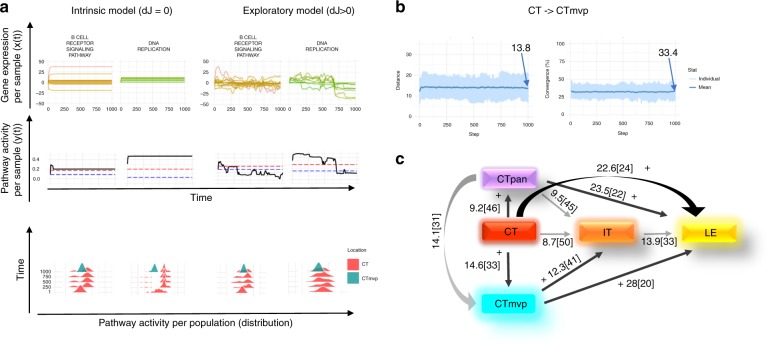
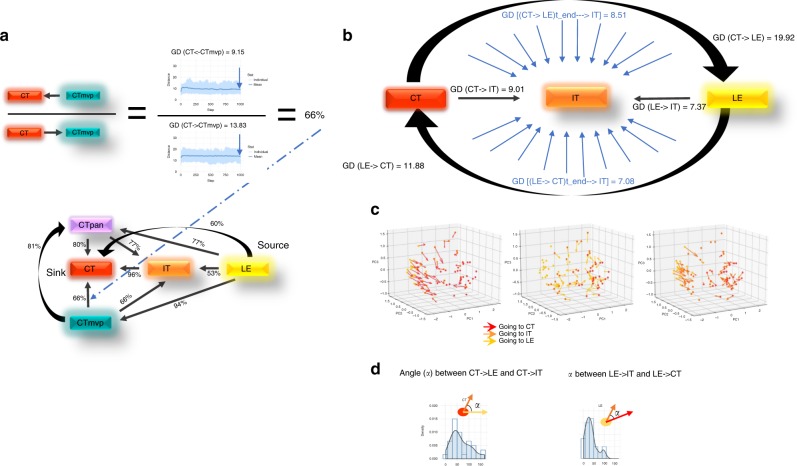
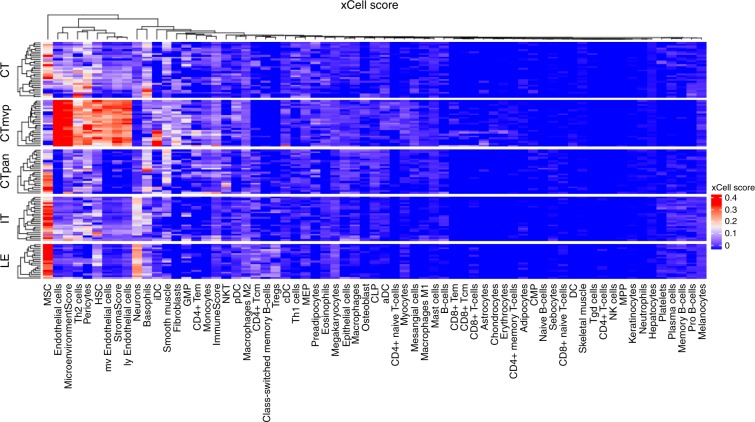
Glioblastoma (GBM) is an aggressive type of brain cancer with remarkable cell migration and adaptation capabilities. Exploratory adaptation-utilization of random changes in gene regulation for adaptive benefits-was recently proposed as the process enabling organisms to survive unforeseen conditions. We investigate whether exploratory adaption explains how GBM cells from different anatomic regions of the tumor cope with micro-environmental pressures. We introduce new notions of phenotype and phenotype distance, and determine probable spatial-phenotypic trajectories based on patient data. While some cell phenotypes are inherently plastic, others are intrinsically rigid with respect to phenotypic transitions. We demonstrate that stochastic exploration of the regulatory network structure confers benefits through enhanced adaptive capacity in new environments. Interestingly, even with exploratory capacity, phenotypic paths are constrained to pass through specific, spatial-phenotypic ranges. This work has important implications for understanding how such adaptation contributes to the recurrence dynamics of GBM and other solid tumors.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Discovering gene-environment interactions in glioblastoma through a comprehensive data integration bioinformatics method.Neurotoxicology. 2013 Mar;35:1-14. doi: 10.1016/j.neuro.2012.11.001. Epub 2012 Dec 20. Neurotoxicology. 2013. PMID: 23261424
-
Comprehensive genetic alteration profiling in primary and recurrent glioblastoma.J Neurooncol. 2019 Mar;142(1):111-118. doi: 10.1007/s11060-018-03070-2. Epub 2018 Dec 9. J Neurooncol. 2019. PMID: 30535594
-
Differential expression of MicroRNAs in patients with glioblastoma after concomitant chemoradiotherapy.OMICS. 2013 May;17(5):259-68. doi: 10.1089/omi.2012.0065. Epub 2013 Apr 15. OMICS. 2013. PMID: 23586679
-
Quantitative Clinical Imaging Methods for Monitoring Intratumoral Evolution.Methods Mol Biol. 2017;1513:61-81. doi: 10.1007/978-1-4939-6539-7_6. Methods Mol Biol. 2017. PMID: 27807831 Review.
-
The role of octamer binding transcription factors in glioblastoma multiforme.Biochim Biophys Acta. 2016 Jun;1859(6):805-11. doi: 10.1016/j.bbagrm.2016.03.003. Epub 2016 Mar 8. Biochim Biophys Acta. 2016. PMID: 26968235 Free PMC article. Review.
Cited by
-
Spatiotemporal dynamics of a glioma immune interaction model.Sci Rep. 2021 Nov 17;11(1):22385. doi: 10.1038/s41598-021-00985-1. Sci Rep. 2021. PMID: 34789751 Free PMC article.
-
Tumor cell plasticity, heterogeneity, and resistance in crucial microenvironmental niches in glioma.Nat Commun. 2021 Feb 12;12(1):1014. doi: 10.1038/s41467-021-21117-3. Nat Commun. 2021. PMID: 33579922 Free PMC article.
-
Cancer progression as a learning process.iScience. 2022 Feb 14;25(3):103924. doi: 10.1016/j.isci.2022.103924. eCollection 2022 Mar 18. iScience. 2022. PMID: 35265809 Free PMC article. Review.
-
Benchmarking algorithms for pathway activity transformation of single-cell RNA-seq data.Comput Struct Biotechnol J. 2020 Oct 15;18:2953-2961. doi: 10.1016/j.csbj.2020.10.007. eCollection 2020. Comput Struct Biotechnol J. 2020. PMID: 33209207 Free PMC article.
-
In-depth analysis of endogenous retrovirus expression in glioblastoma.Mob DNA. 2025 Jul 16;16(1):29. doi: 10.1186/s13100-025-00365-w. Mob DNA. 2025. PMID: 40671039 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
