

Diagnostic yield of panel-based genetic testing in syndromic inherited retinal disease
- PMID: 31836858
- PMCID: PMC7171123
- DOI: 10.1038/s41431-019-0548-5
Diagnostic yield of panel-based genetic testing in syndromic inherited retinal disease
Abstract
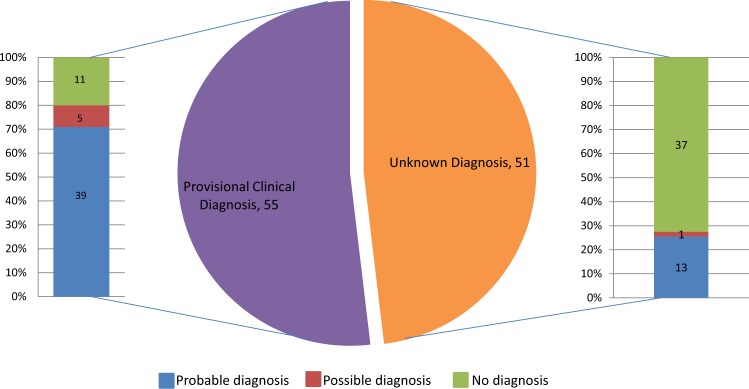
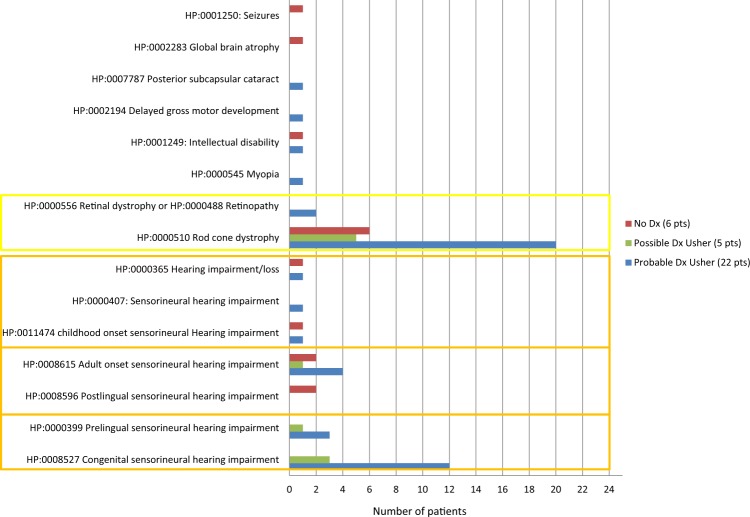
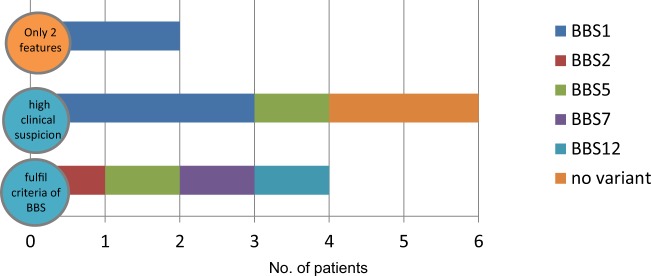
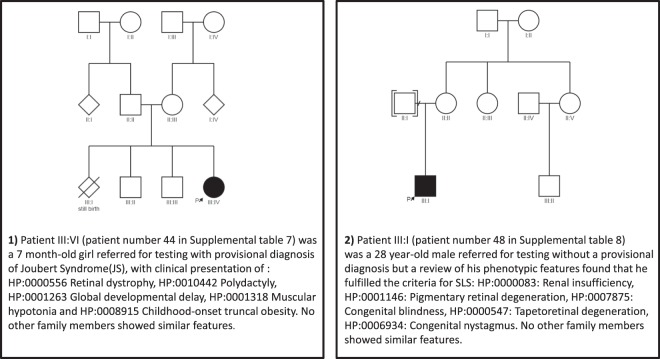
Thirty percent of all inherited retinal disease (IRD) is accounted for by conditions with extra-ocular features. This study aimed to establish the genetic diagnostic pick-up rate for IRD patients with one or more extra-ocular features undergoing panel-based screening in a clinical setting. One hundred and six participants, tested on a gene panel which contained both isolated and syndromic IRD genes, were retrospectively ascertained from the Manchester Genomic Diagnostics Laboratory database spanning 6 years (2012-2017). Phenotypic features were extracted from the clinical notes and classified according to Human Phenotype Ontology; all identified genetic variants were interpreted in accordance to the American College of Medical Genetics and Genomics guidelines. Overall, 49% (n = 52) of patients received a probable genetic diagnosis. A further 6% (n = 6) had a single disease-associated variant in an autosomal recessive disease-relevant gene. Fifty-two percent (n = 55) of patients had a clinical diagnosis at the time of testing. Of these, 71% (n = 39) received a probable genetic diagnosis. By contrast, for those without a provisional clinical diagnosis (n = 51), only 25% (n = 13) received a probable genetic diagnosis. The clinical diagnosis of Usher (n = 33) and Bardet-Biedl syndrome (n = 10) was confirmed in 67% (n = 22) and 80% (n = 8), respectively. The testing diagnostic rate in patients with clinically diagnosed multisystemic IRD conditions was significantly higher than those without one (71% versus 25%; p value < 0.001). The lower pick-up rate in patients without a clinical diagnosis suggests that panel-based approaches are unlikely to be the most effective means of achieving a molecular diagnosis for this group. Here, we suggest that genome-wide approaches (whole exome or genome) are more appropriate.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
