

Towards personalized treatment for early stage HER2-positive breast cancer
- PMID: 31836877
- PMCID: PMC8023395
- DOI: 10.1038/s41571-019-0299-9
Towards personalized treatment for early stage HER2-positive breast cancer
Abstract
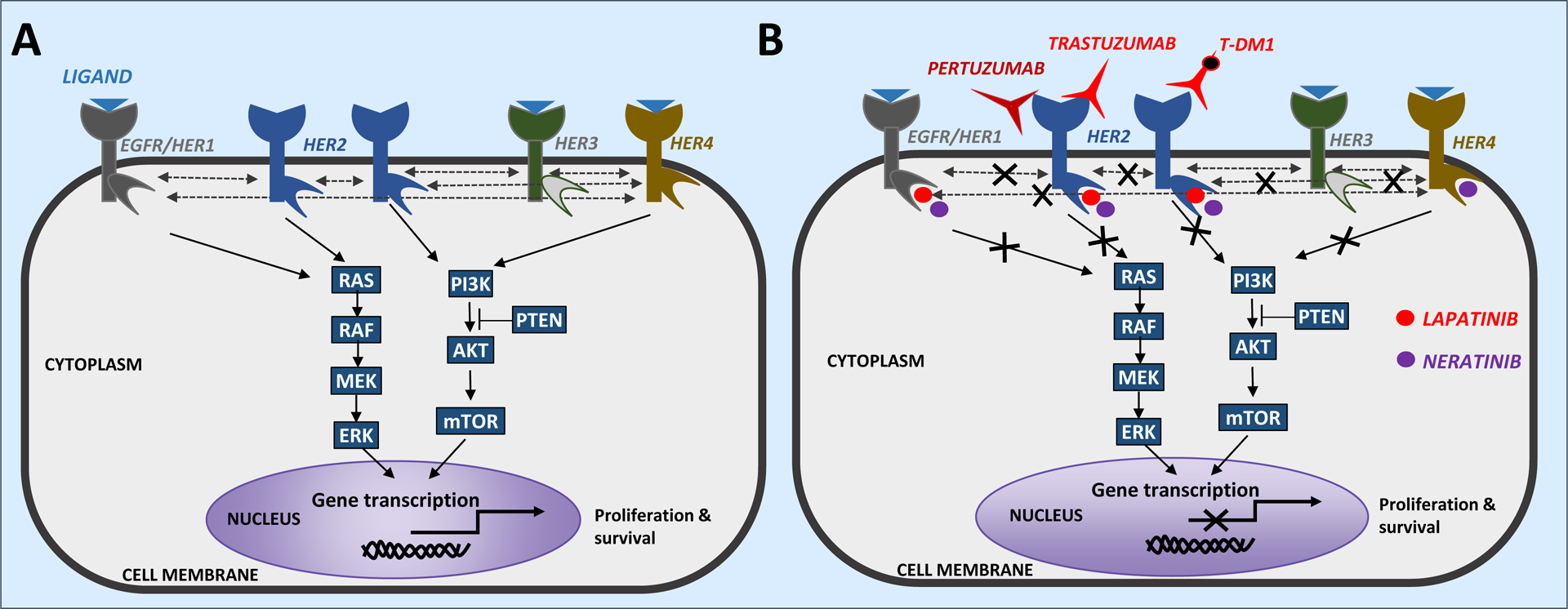
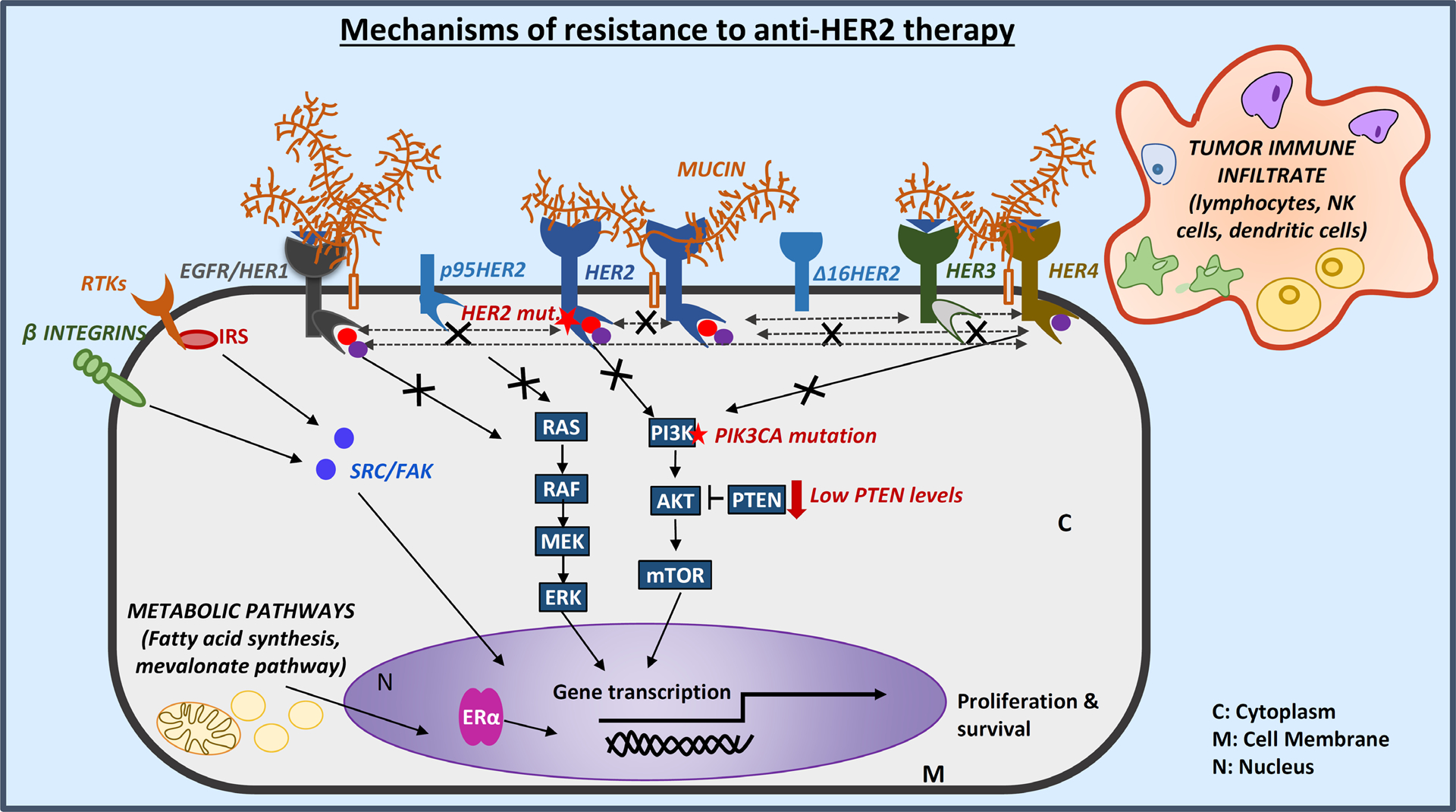
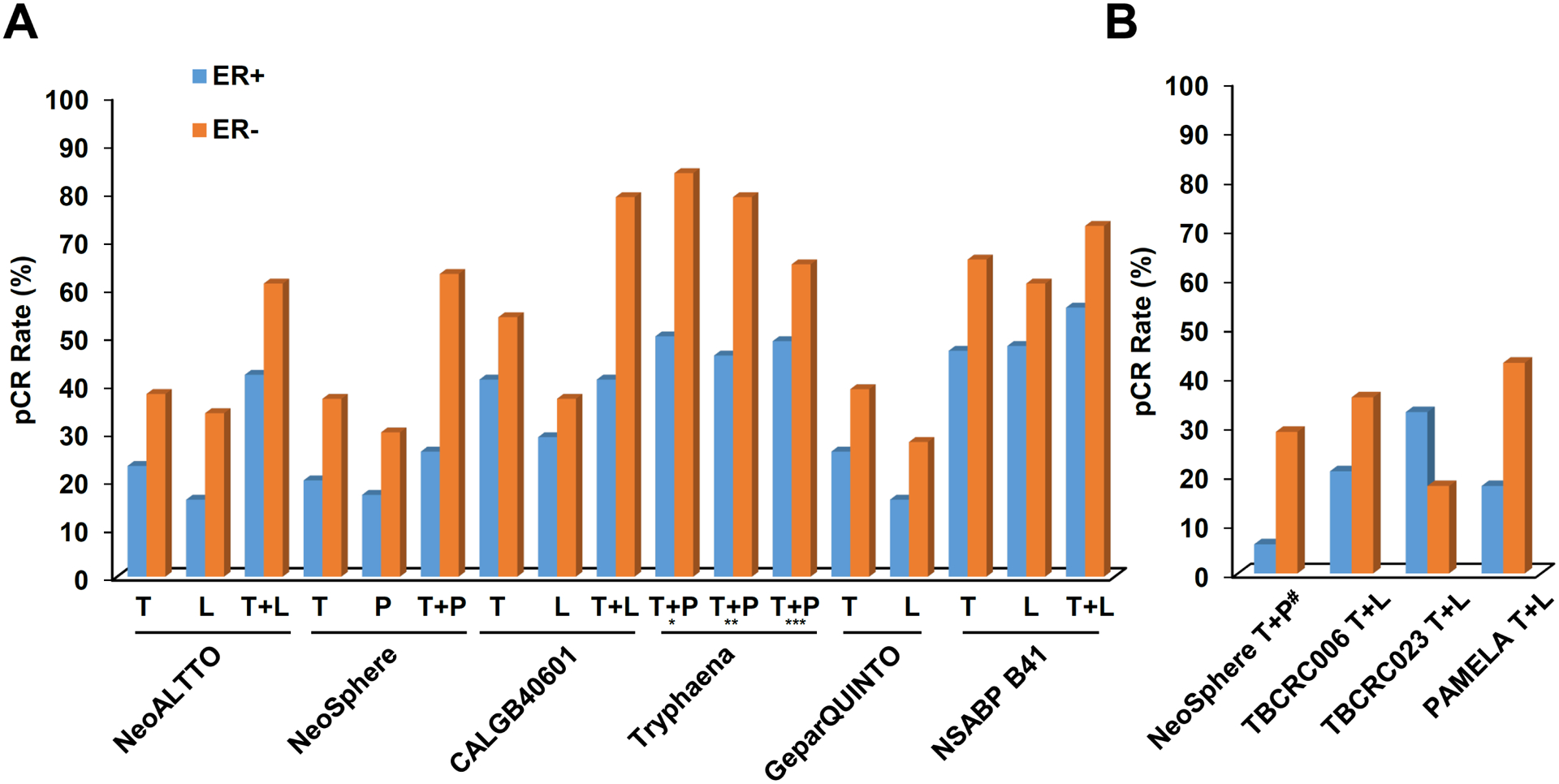
Advances in HER2-targeted therapies have improved the survival of patients with HER2-positive breast cancer. The standard-of-care treatment for localized disease has been chemotherapy and 1 year of adjuvant HER2-targeted therapy, typically with the anti-HER2 antibody trastuzumab. Despite the effectiveness of this treatment, disease relapse occurs in a subset of patients; thus, focus has been placed on escalating treatment by either combining different HER2-targeted agents or extending the duration of HER2-targeted therapy. Indeed, dual HER2-targeted therapies and extended-duration anti-HER2 therapy, as well as adjuvant therapy with the anti-HER2 antibody-drug conjugate T-DM1, have all been approved for clinical use. Emerging evidence suggests, however, that some patients do not derive sufficient benefit from these additional therapies to offset the associated toxicities and/or costs. Similarly, the universal use of chemotherapy might not benefit all patients, and treatment de-escalation through omission of chemotherapy has shown promise in clinical trials and is currently being explored further. The future of precision medicine should therefore involve tailoring of therapy based on the genetics and biology of each tumour and the clinical characteristics of each patient. Predictive biomarkers that enable the identification of patients who will benefit from either escalated or de-escalated treatment will be crucial to this approach. In this Review, we summarize the available HER2-targeted agents and associated mechanisms of resistance, and describe the current therapeutic landscape of early stage HER2-positive breast cancer, focusing on strategies for treatment escalation or de-escalation.
Conflict of interest statement
Competing interests
C.K.O. has received research funding from AstraZeneca and GlaxoSmithKline, has served on advisory boards for AstraZeneca, Genentech and Tolmar Pharmaceuticals, has been a data monitoring committee member for Eli Lilly and is a stockholder of GeneTex. M.F.R. receives research support from GSK via his institution and has been a consultant for Daiichi, Genentech, Macrogenics and Novartis. R.S. has received research funding from AstraZeneca, Gilead Sciences, GlaxoSmithKline and PUMA Biotechnology, and has been a consultant or advisory committee member for Eli Lilly and Macrogenics. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
