

FDA oversight of NSIGHT genomic research: the need for an integrated systems approach to regulation
- PMID: 31839987
- PMCID: PMC6904743
- DOI: 10.1038/s41525-019-0105-8
FDA oversight of NSIGHT genomic research: the need for an integrated systems approach to regulation
Abstract
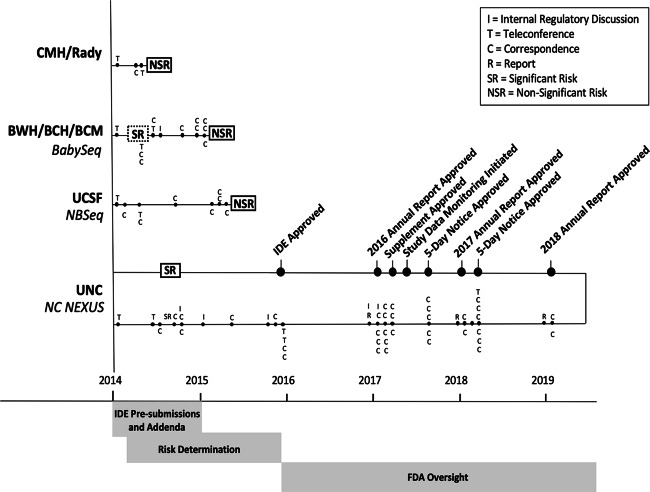
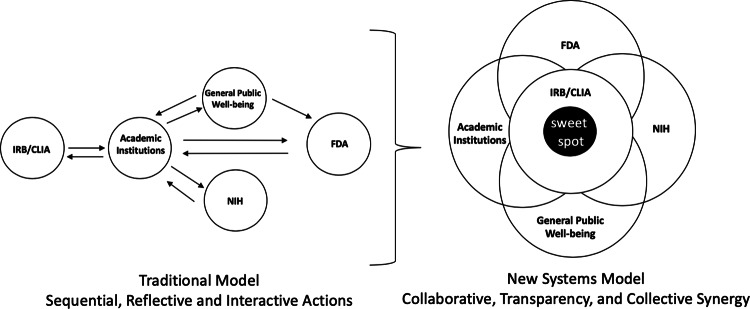
The National Institutes of Health (NIH) funded the Newborn Sequencing In Genomic medicine and public HealTh (NSIGHT) Consortium to investigate the implications, challenges, and opportunities associated with the possible use of genomic sequence information in the newborn period. Following announcement of the NSIGHT awardees in 2013, the Food and Drug Administration (FDA) contacted investigators and requested that pre-submissions to investigational device exemptions (IDE) be submitted for the use of genomic sequencing under Title 21 of the Code of Federal Regulations (21 CFR) part 812. IDE regulation permits clinical investigation of medical devices that have not been approved by the FDA. To our knowledge, this marked the first time the FDA determined that NIH-funded clinical genomic research projects are subject to IDE regulation. Here, we review the history of and rationale behind FDA oversight of clinical research and the NSIGHT Consortium's experiences in navigating the IDE process. Overall, NSIGHT investigators found that FDA's application of existing IDE regulations and medical device definitions aligned imprecisely with the aims of publicly funded exploratory clinical research protocols. IDE risk assessments by the FDA were similar to, but distinct from, protocol risk assessments conducted by local Institutional Review Boards (IRBs), and had the potential to reflect novel oversight of emerging genomic technologies. However, the pre-IDE and IDE process delayed the start of NSIGHT research studies by an average of 10 months, and significantly limited the scope of investigation in two of the four NIH approved projects. Based on the experience of the NSIGHT Consortium, we conclude that policies and practices governing the development and use of novel genomic technologies in clinical research urgently need clarification in order to mitigate potentially conflicting or redundant oversight by IRBs, NIH, FDA, and state authorities.
Keywords: Genetics research; Health policy; Next-generation sequencing; Paediatric research; Population screening.
© The Author(s) 2019.
Conflict of interest statement
Competing interestsL.V.M., A.M.B., P.B.A. I.A.H., F.C., J.S.B., R.B.P., C.M.P., K.C, I.A.H., A.H.B., and S.F.K. declare no competing interests. S.E.B. received research support at UC Berkeley from TATA Consultancy Services.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
