

Etiology, Risk Factors, and Biomarkers in Systemic Sclerosis with Interstitial Lung Disease
- PMID: 31841044
- PMCID: PMC7068837
- DOI: 10.1164/rccm.201903-0563CI
Etiology, Risk Factors, and Biomarkers in Systemic Sclerosis with Interstitial Lung Disease
Abstract
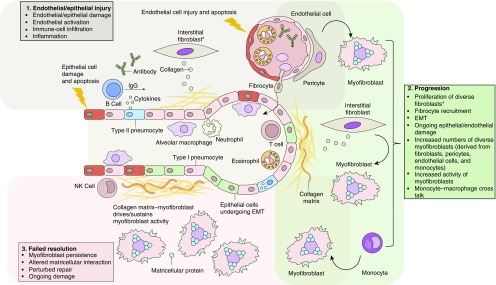
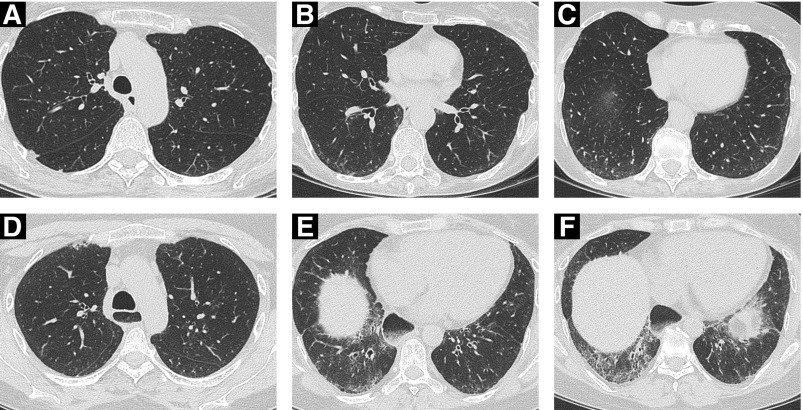
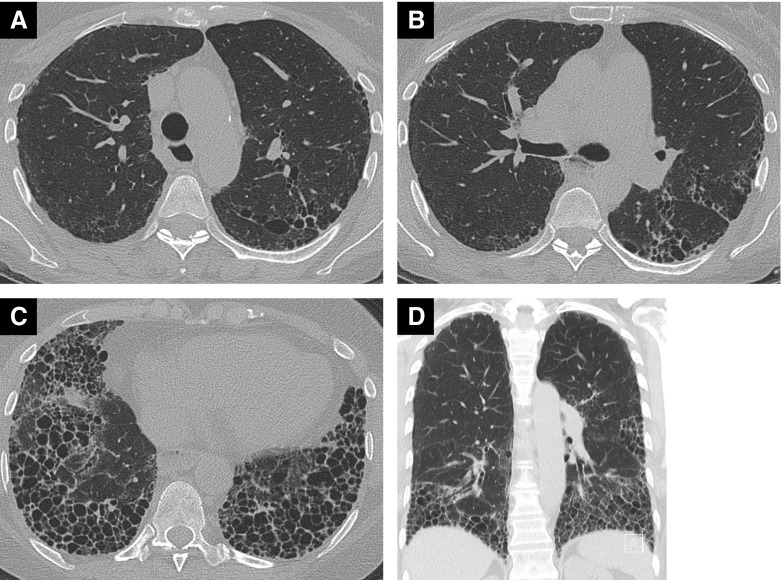
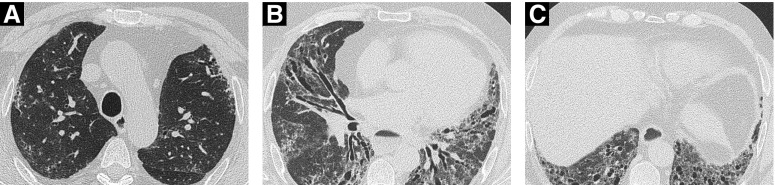
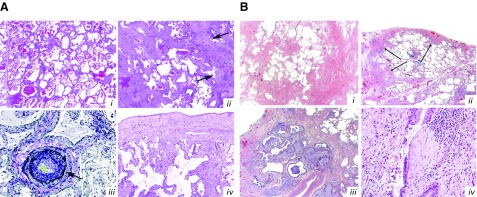
Systemic sclerosis (SSc) is a complex, multiorgan, autoimmune disease. Lung fibrosis occurs in ∼80% of patients with SSc; 25% to 30% develop progressive interstitial lung disease (ILD). The pathogenesis of fibrosis in SSc-associated ILD (SSc-ILD) involves cellular injury, activation/differentiation of mesenchymal cells, and morphological/biological changes in epithelial/endothelial cells. Risk factors for progressive SSc-ILD include older age, male sex, degree of lung involvement on baseline high-resolution computed tomography imaging, reduced DlCO, and reduced FVC. SSc-ILD does not share the genetic risk architecture observed in idiopathic pulmonary fibrosis (IPF), with key risk factors yet to be identified. Presence of anti-Scl-70 antibodies and absence of anti-centromere antibodies indicate increased likelihood of progressive ILD. Elevated levels of serum Krebs von den Lungen-6 and C-reactive protein are both associated with SSc-ILD severity and predict SSc-ILD progression. A promising prognostic indicator is serum chemokine (C-C motif) ligand 18. SSc-ILD shares similarities with IPF, although clear differences exist. Histologically, a nonspecific interstitial pneumonia pattern is commonly observed in SSc-ILD, whereas IPF is defined by usual interstitial pneumonia. The course of SSc-ILD is variable, ranging from minor, stable disease to a progressive course, whereas all patients with IPF experience progression of disease. Although appropriately treated patients with SSc-ILD have better chances of stabilization and survival, a relentlessly progressive course, akin to IPF, is seen in a minority. Better understanding of cellular and molecular pathogenesis, genetic risk, and distinctive features of SSc-ILD and identification of robust prognostic biomarkers are needed for optimal disease management.
Keywords: autoimmune diseases; biomarkers; interstitial lung diseases; risk factors; systemic sclerosis.
Figures
References
-
- Denton CP, Hughes M, Gak N, Vila J, Buch MH, Chakravarty K, et al. BSR and BHPR Standards, Guidelines and Audit Working Group. BSR and BHPR guideline for the treatment of systemic sclerosis. Rheumatology (Oxford) 2016;55:1906–1910. - PubMed
-
- Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390:1685–1699. - PubMed
-
- Ranque B, Mouthon L. Geoepidemiology of systemic sclerosis. Autoimmun Rev. 2010;9:A311–A318. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
