

PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis
- PMID: 31841108
- PMCID: PMC7299340
- DOI: 10.7554/eLife.52135
PCK1 and DHODH drive colorectal cancer liver metastatic colonization and hypoxic growth by promoting nucleotide synthesis
Abstract
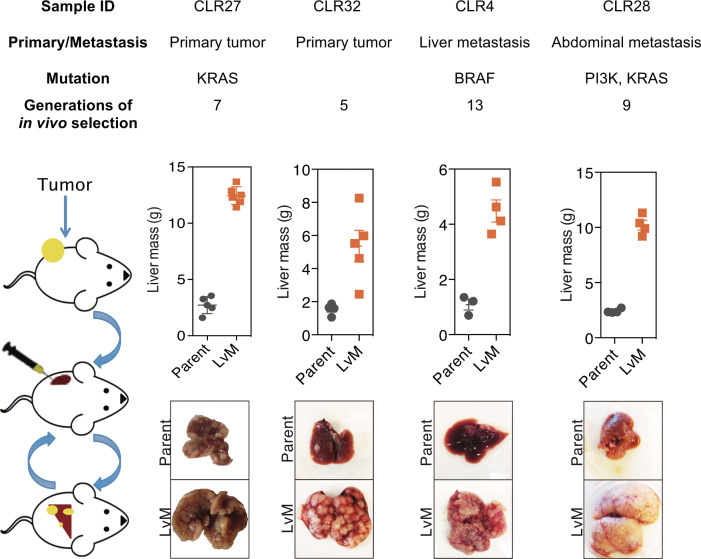
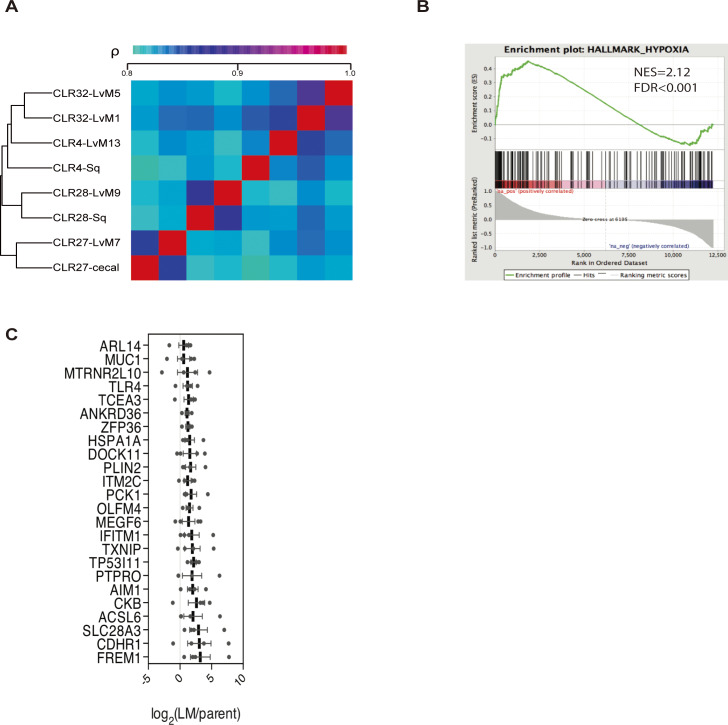
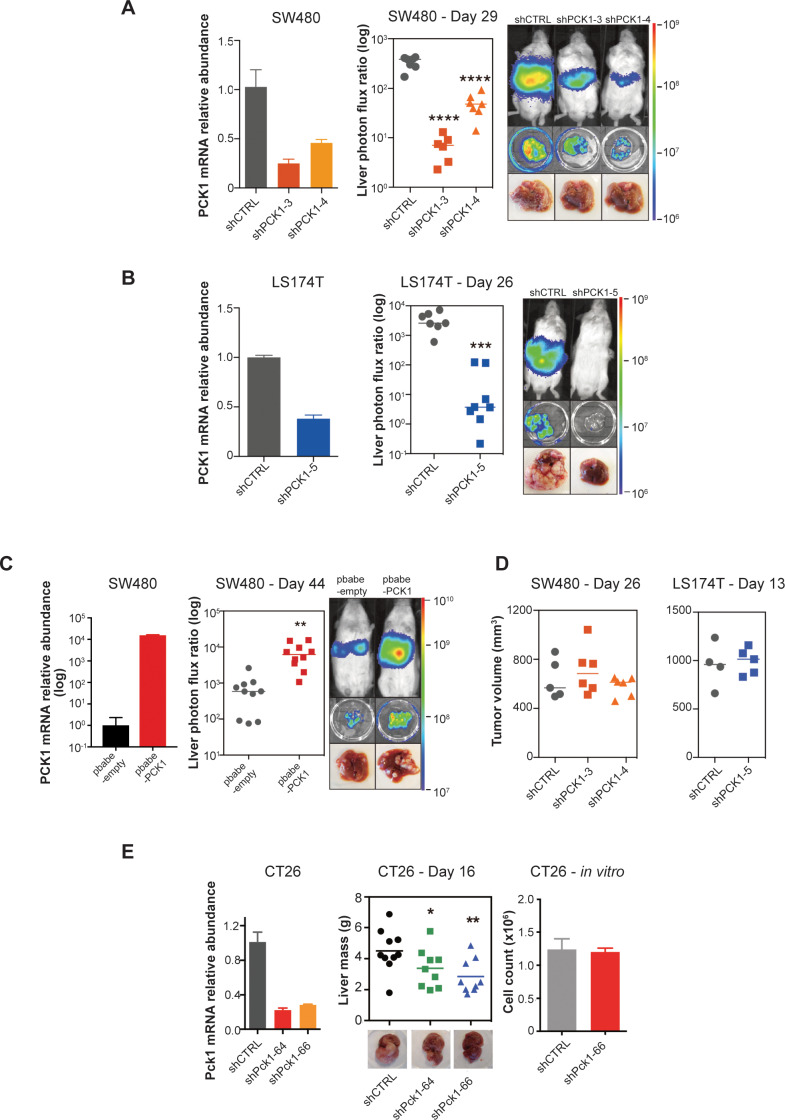
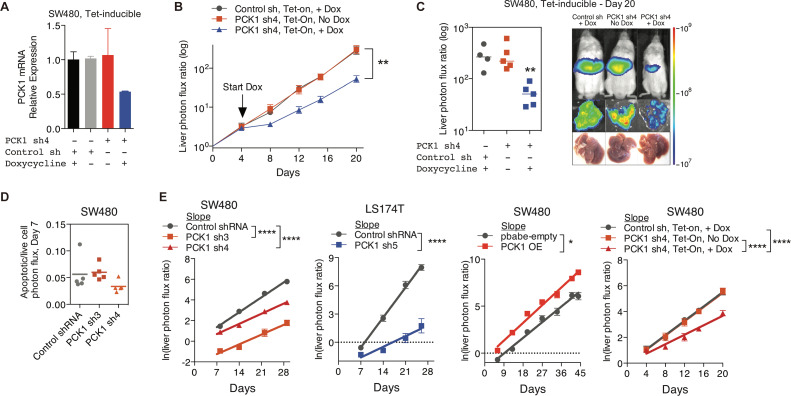
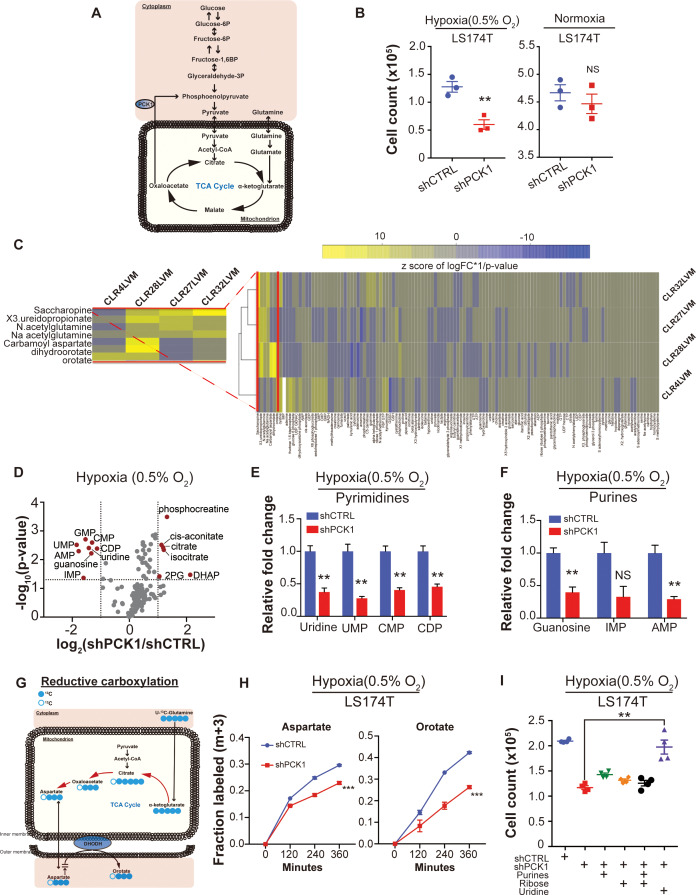
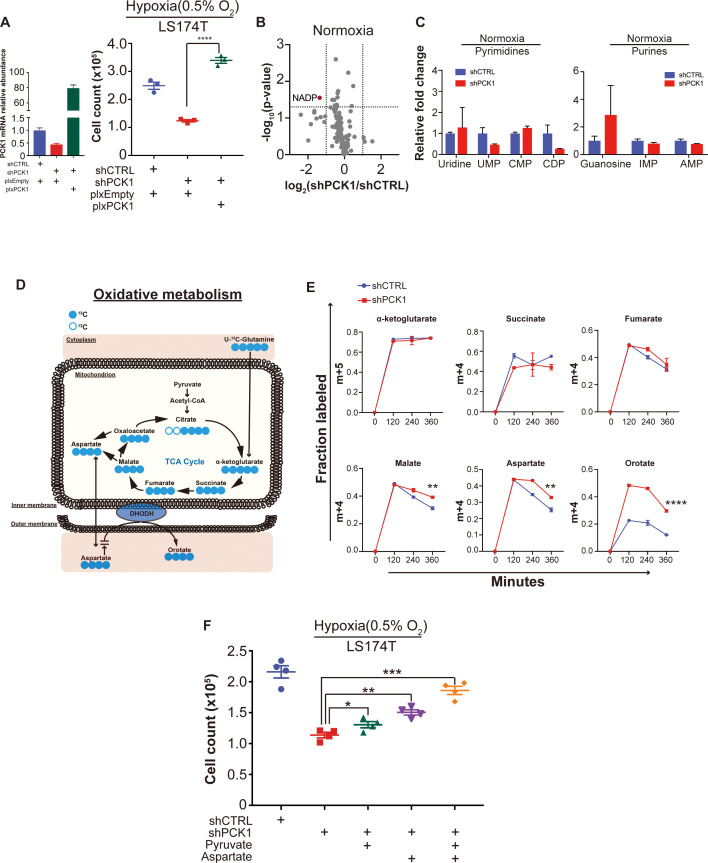
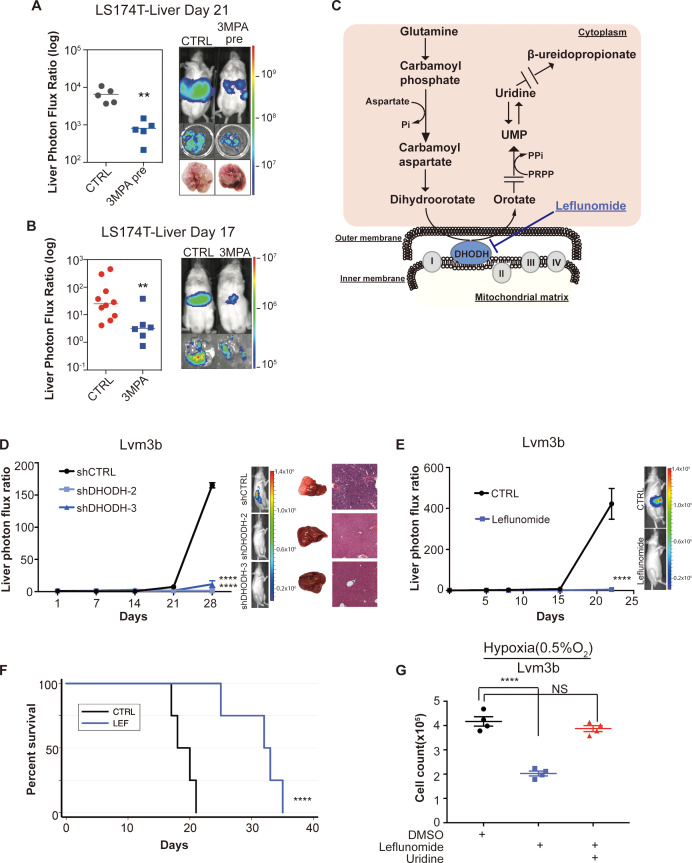
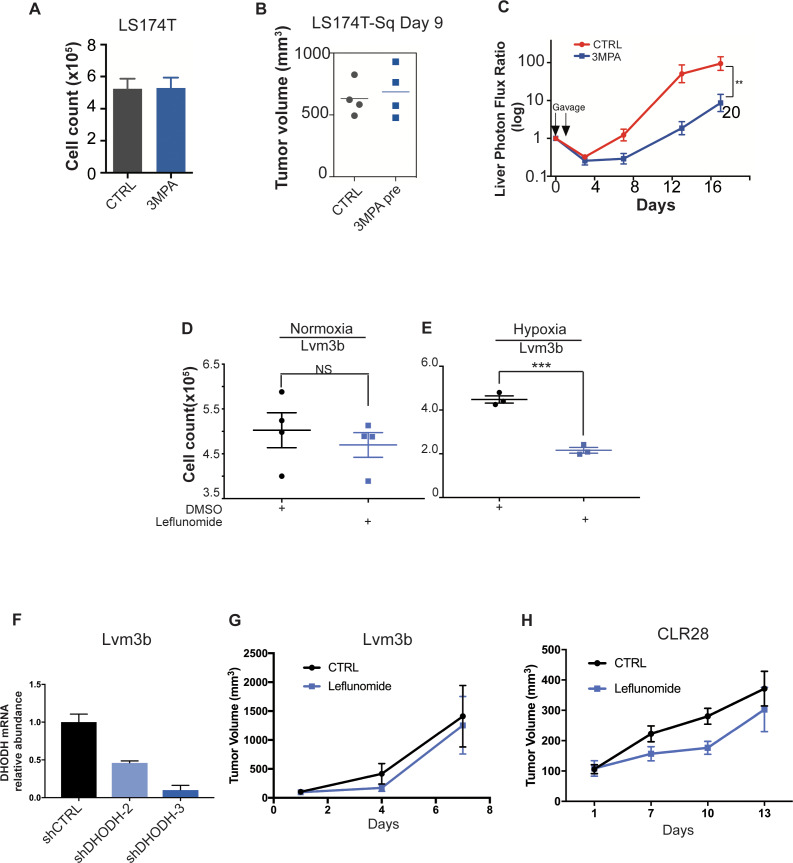
Colorectal cancer (CRC) is a major cause of human death. Mortality is primarily due to metastatic organ colonization, with the liver being the main organ affected. We modeled metastatic CRC (mCRC) liver colonization using patient-derived primary and metastatic tumor xenografts (PDX). Such PDX modeling predicted patient survival outcomes. In vivo selection of multiple PDXs for enhanced metastatic colonization capacity upregulated the gluconeogenic enzyme PCK1, which enhanced liver metastatic growth by driving pyrimidine nucleotide biosynthesis under hypoxia. Consistently, highly metastatic tumors upregulated multiple pyrimidine biosynthesis intermediary metabolites. Therapeutic inhibition of the pyrimidine biosynthetic enzyme DHODH with leflunomide substantially impaired CRC liver metastatic colonization and hypoxic growth. Our findings provide a potential mechanistic basis for the epidemiologic association of anti-gluconeogenic drugs with improved CRC metastasis outcomes, reveal the exploitation of a gluconeogenesis enzyme for pyrimidine biosynthesis under hypoxia, and implicate DHODH and PCK1 as metabolic therapeutic targets in CRC metastatic progression.
Keywords: cancer biology; cancer metabolism; cancer metastasis; human; human biology; medicine; mouse.
© 2019, Yamaguchi et al.
Conflict of interest statement
NY, EW, AN, ML, HG, YJ, PP, LS, TK, JL, Ed, ST No competing interests declared
Figures

References
-
- Bajzikova M, Kovarova J, Coelho AR, Boukalova S, Oh S, Rohlenova K, Svec D, Hubackova S, Endaya B, Judasova K, Bezawork-Geleta A, Kluckova K, Chatre L, Zobalova R, Novakova A, Vanova K, Ezrova Z, Maghzal GJ, Magalhaes Novais S, Olsinova M, Krobova L, An YJ, Davidova E, Nahacka Z, Sobol M, Cunha-Oliveira T, Sandoval-Acuña C, Strnad H, Zhang T, Huynh T, Serafim TL, Hozak P, Sardao VA, Koopman WJH, Ricchetti M, Oliveira PJ, Kolar F, Kubista M, Truksa J, Dvorakova-Hortova K, Pacak K, Gurlich R, Stocker R, Zhou Y, Berridge MV, Park S, Dong L, Rohlena J, Neuzil J. Reactivation of dihydroorotate Dehydrogenase-Driven pyrimidine biosynthesis restores tumor growth of Respiration-Deficient Cancer cells. Cell Metabolism. 2019;29:399–416. doi: 10.1016/j.cmet.2018.10.014. - DOI - PMC - PubMed
-
- Chandel NS. Navigating Metabolism. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory Press; 2015.
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
