

A Molecular Revolution in the Treatment of Hemophilia
- PMID: 31843450
- PMCID: PMC7132613
- DOI: 10.1016/j.ymthe.2019.11.006
A Molecular Revolution in the Treatment of Hemophilia
Abstract
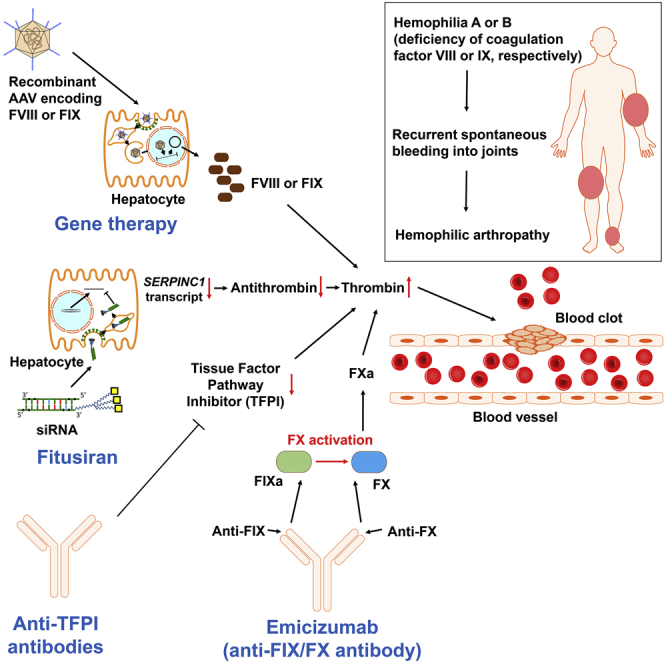
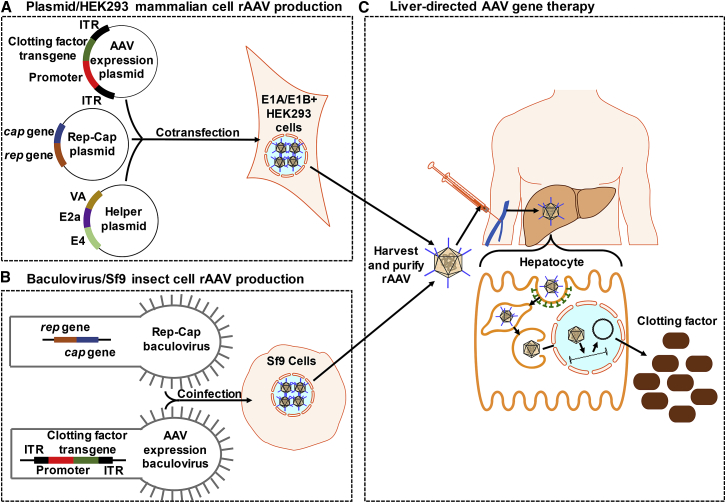
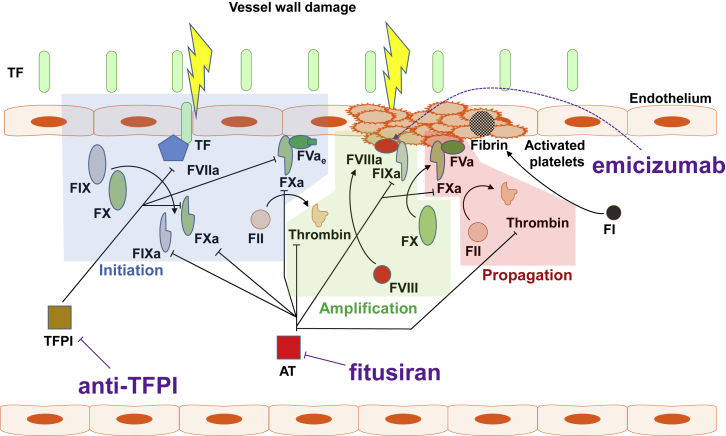
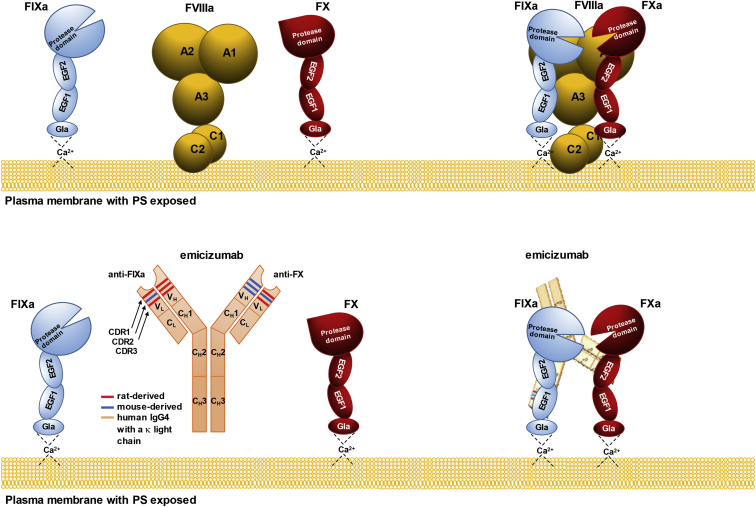
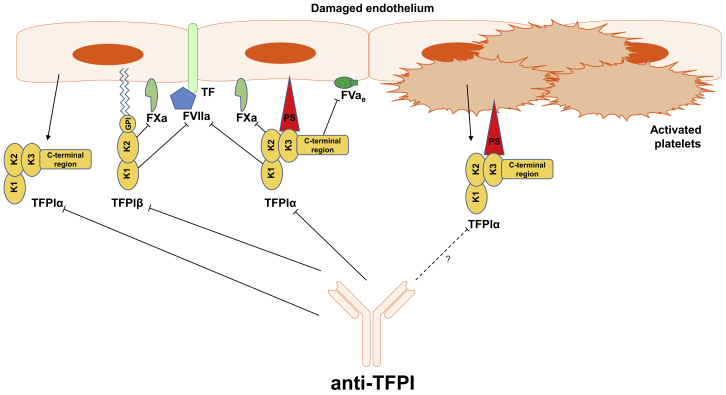
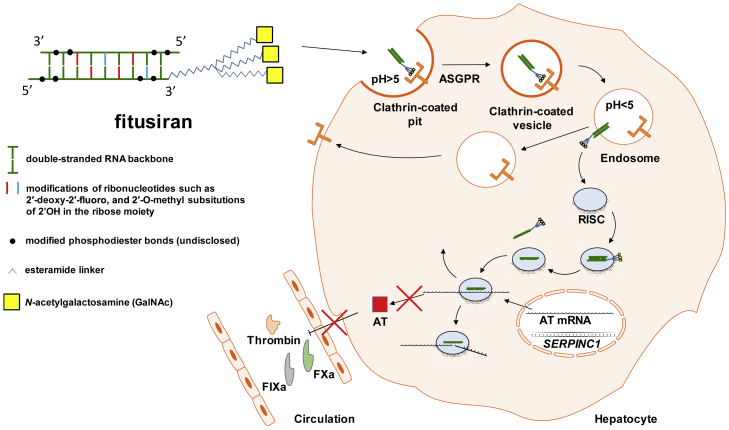
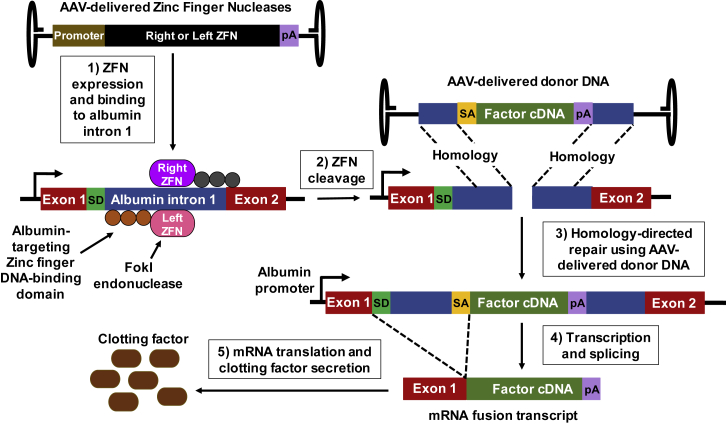
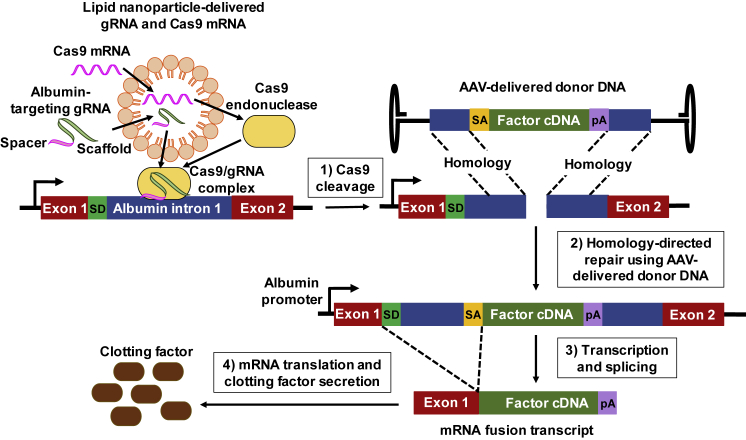
For decades, the monogenetic bleeding disorders hemophilia A and B (coagulation factor VIII and IX deficiency) have been treated with systemic protein replacement therapy. Now, diverse molecular medicines, ranging from antibody to gene to RNA therapy, are transforming treatment. Traditional replacement therapy requires twice to thrice weekly intravenous infusions of factor. While extended half-life products may reduce the frequency of injections, patients continue to face a lifelong burden of the therapy, suboptimal protection from bleeding and joint damage, and potential development of neutralizing anti-drug antibodies (inhibitors) that require less efficacious bypassing agents and further reduce quality of life. Novel non-replacement and gene therapies aim to address these remaining issues. A recently approved factor VIII-mimetic antibody accomplishes hemostatic correction in patients both with and without inhibitors. Antibodies against tissue factor pathway inhibitor (TFPI) and antithrombin-specific small interfering RNA (siRNA) target natural anticoagulant pathways to rebalance hemostasis. Adeno-associated virus (AAV) gene therapy provides lasting clotting factor replacement and can also be used to induce immune tolerance. Multiple gene-editing techniques are under clinical or preclinical investigation. Here, we provide a comprehensive overview of these approaches, explain how they differ from standard therapies, and predict how the hemophilia treatment landscape will be reshaped.
Copyright © 2019 The American Society of Gene and Cell Therapy. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- World Federation of Hemophilia . World Federation of Hemophilia; 2017. Report of the Annual Global Survey 2016.http://www1.wfh.org/publications/files/pdf-1690.pdf
-
- Srivastava A., Brewer A.K., Mauser-Bunschoten E.P., Key N.S., Kitchen S., Llinas A., Ludlam C.A., Mahlangu J.N., Mulder K., Poon M.C., Street A., Treatment Guidelines Working Group on Behalf of the World Federation of Hemophilia Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–e47. - PubMed
-
- Mannucci P.M., Tuddenham E.G.D. The hemophilias—from royal genes to gene therapy. N. Engl. J. Med. 2001;344:1773–1779. - PubMed
-
- Stonebraker J.S., Bolton-Maggs P.H.B., Soucie J.M., Walker I., Brooker M. A study of variations in the reported haemophilia A prevalence around the world. Haemophilia. 2010;16:20–32. - PubMed
-
- Schrijvers L.H., Schuurmans M.J., Fischer K. Promoting self-management and adherence during prophylaxis: evidence-based recommendations for haemophilia professionals. Haemophilia. 2016;22:499–506. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
