

Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa, Asia and Oceania
- PMID: 31844061
- PMCID: PMC6914791
- DOI: 10.1038/s41467-019-13480-z
Insights into malaria susceptibility using genome-wide data on 17,000 individuals from Africa, Asia and Oceania
Abstract
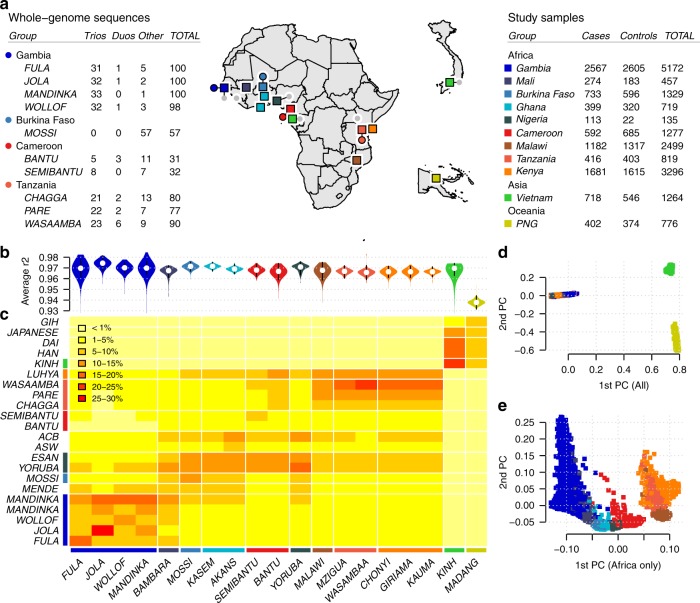
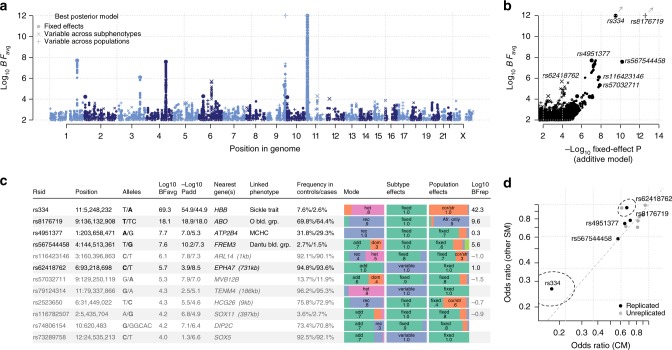
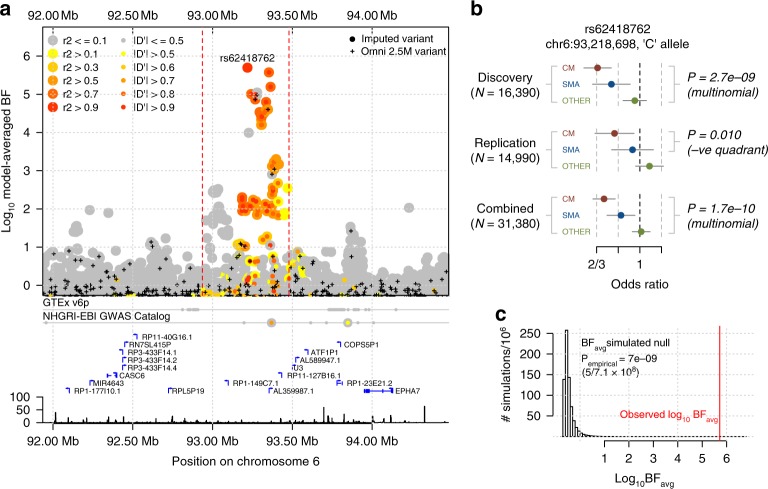
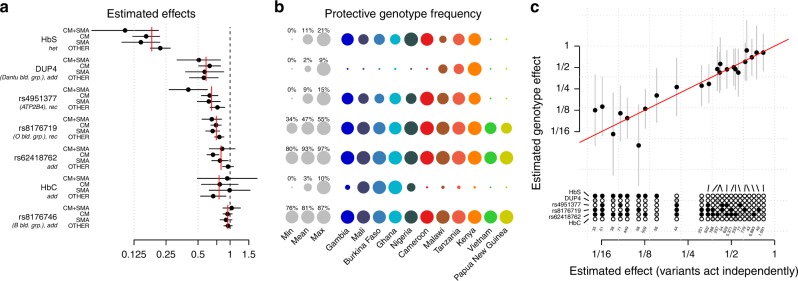
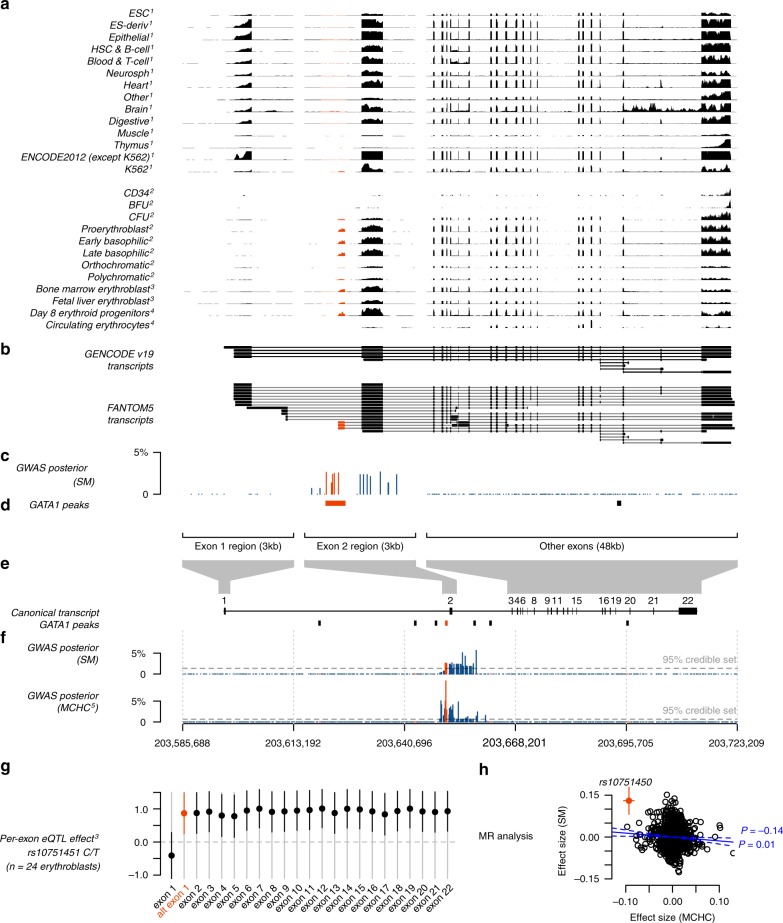
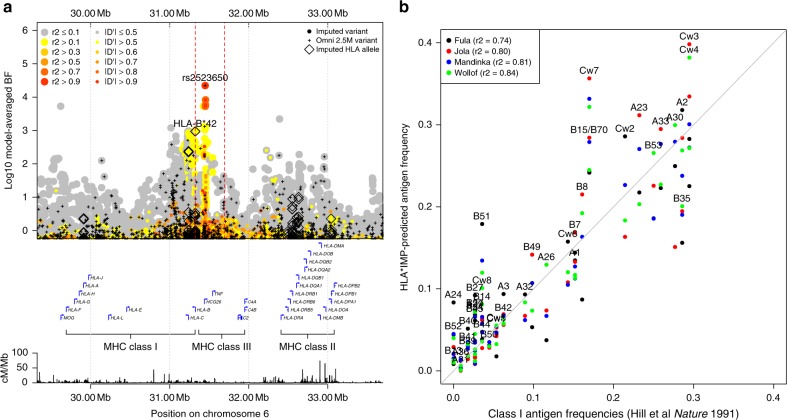
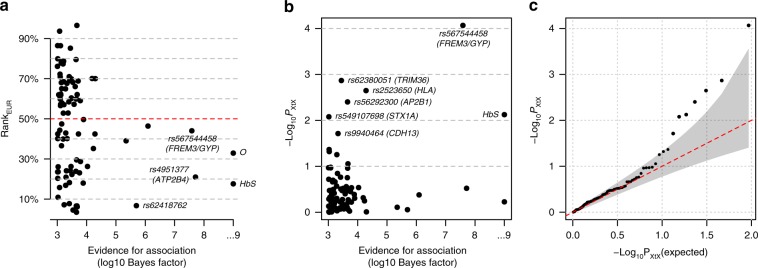
The human genetic factors that affect resistance to infectious disease are poorly understood. Here we report a genome-wide association study in 17,000 severe malaria cases and population controls from 11 countries, informed by sequencing of family trios and by direct typing of candidate loci in an additional 15,000 samples. We identify five replicable associations with genome-wide levels of evidence including a newly implicated variant on chromosome 6. Jointly, these variants account for around one-tenth of the heritability of severe malaria, which we estimate as ~23% using genome-wide genotypes. We interrogate available functional data and discover an erythroid-specific transcription start site underlying the known association in ATP2B4, but are unable to identify a likely causal mechanism at the chromosome 6 locus. Previously reported HLA associations do not replicate in these samples. This large dataset will provide a foundation for further research on thegenetic determinants of malaria resistance in diverse populations.
Conflict of interest statement
C.C.A.S. is a shareholder in, and current employee of, Genomics PLC.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
