

A large peptidome dataset improves HLA class I epitope prediction across most of the human population
- PMID: 31844290
- PMCID: PMC7008090
- DOI: 10.1038/s41587-019-0322-9
A large peptidome dataset improves HLA class I epitope prediction across most of the human population
Abstract
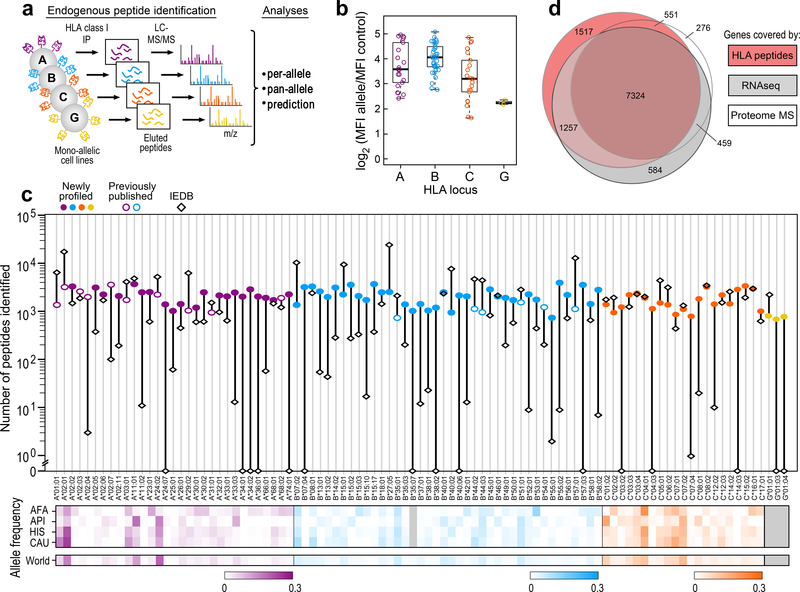
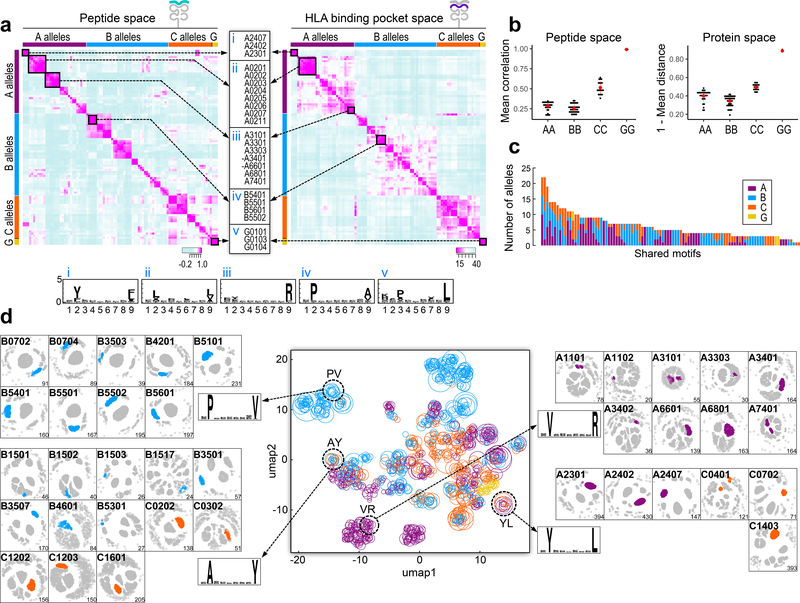
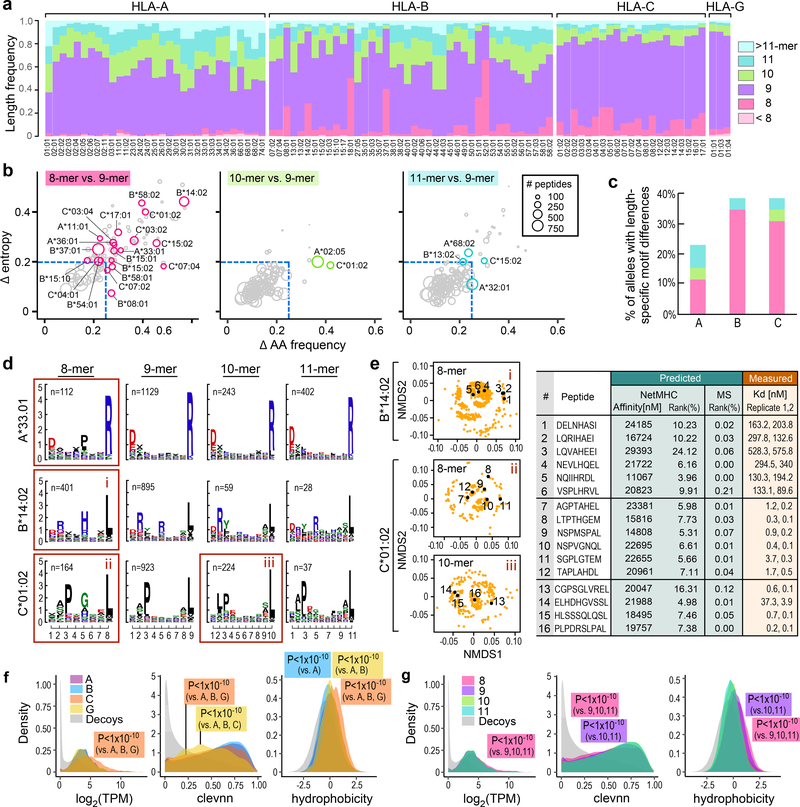
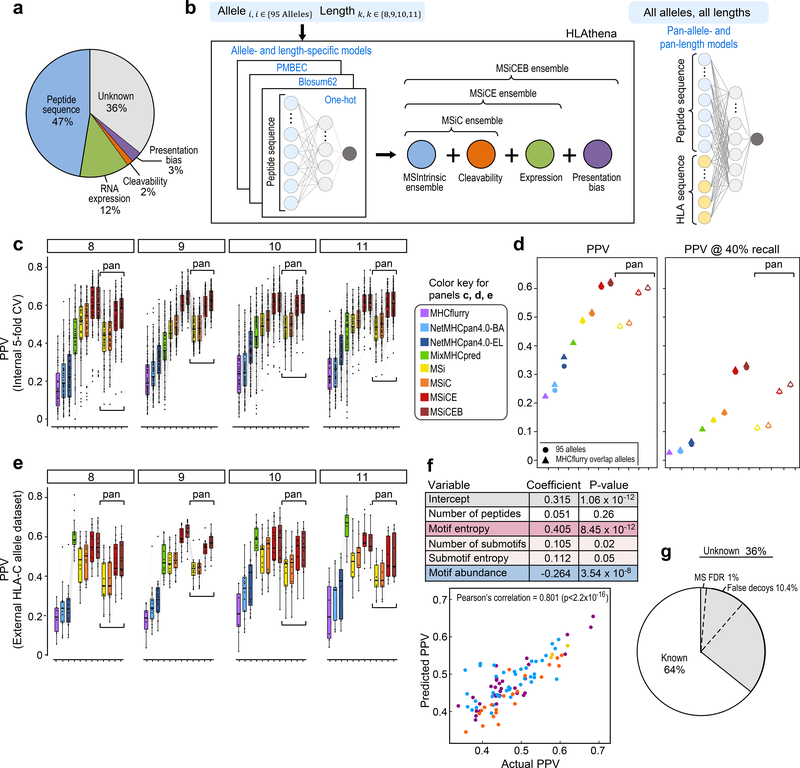
Prediction of HLA epitopes is important for the development of cancer immunotherapies and vaccines. However, current prediction algorithms have limited predictive power, in part because they were not trained on high-quality epitope datasets covering a broad range of HLA alleles. To enable prediction of endogenous HLA class I-associated peptides across a large fraction of the human population, we used mass spectrometry to profile >185,000 peptides eluted from 95 HLA-A, -B, -C and -G mono-allelic cell lines. We identified canonical peptide motifs per HLA allele, unique and shared binding submotifs across alleles and distinct motifs associated with different peptide lengths. By integrating these data with transcript abundance and peptide processing, we developed HLAthena, providing allele-and-length-specific and pan-allele-pan-length prediction models for endogenous peptide presentation. These models predicted endogenous HLA class I-associated ligands with 1.5-fold improvement in positive predictive value compared with existing tools and correctly identified >75% of HLA-bound peptides that were observed experimentally in 11 patient-derived tumor cell lines.
Conflict of interest statement
Competing Interests
D.B.K. has previously advised Neon Therapeutics, and owns equity in Aduro Biotech, Agenus Inc., Armata pharmaceuticals, Biomarin Pharmaceutical Inc., Bristol Myers Squibb Com., Celldex Therapeutics Inc., Editas Medicine Inc., Exelixis Inc., Gilead Sciences Inc., IMV Inc., Lexicon Pharmaceuticals Inc., and Stemline Therapeutics Inc. D.A.B. has received consulting fees from Octane Global, Defined Health, Dedham Group, Adept Field Solutions, Slingshot Insights, Blueprint Partnership, Charles River Associates, Trinity Group, Insight Strategy, and is a member of the RCC translational medicine advisory broad of Bristol-Myers Squibb. K.L.L. owns equity and is a founder of Travera LLC and is an advisor to Bristol Myers Squibb Com. and Rarecyte. S.A.C is a member of the scientific advisory boards of Kymera, PTM BioLabs and BioAnalytix and a scientific advisor to Pfizer and Biogen. C.J.W. and N.H. are founders of Neon Therapeutics and members of its scientific advisory board. N.H. is also an advisor for IFM therapeutics. W.J.L is a member of the scientific advisory board of CareDx. All other authors have no competing interests.
Figures


Comment in
-
Evaluating NetMHCpan performance on non-European HLA alleles not present in training data.Front Immunol. 2024 Jan 16;14:1288105. doi: 10.3389/fimmu.2023.1288105. eCollection 2023. Front Immunol. 2024. PMID: 38292493 Free PMC article.
References
-
- O’Donnell TJ et al. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst 7, 129–132.e4 (2018). - PubMed
Online Methods-only references
-
- Kidera A, Konishi Y, Oka M, Ooi T & Scheraga HA Statistical analysis of the physical properties of the 20 naturally occurring amino acids. J. Protein Chem 4, 23–55 (1985).
-
- McInnes L, Healy J & Melville J UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv [stat.ML] (2018).
-
- Harndahl M et al. Peptide binding to HLA class I molecules: homogenous, high-throughput screening, and affinity assays. J. Biomol. Screen 14, 173–180 (2009). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- P01 CA229092/CA/NCI NIH HHS/United States
- P50 CA101942/CA/NCI NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- T32 CA009172/CA/NCI NIH HHS/United States
- U24 CA224331/CA/NCI NIH HHS/United States
- R21 CA216772/CA/NCI NIH HHS/United States
- R01 CA155010/CA/NCI NIH HHS/United States
- U01 CA214125/CA/NCI NIH HHS/United States
- R01 CA279391/CA/NCI NIH HHS/United States
- T32 CA207021/CA/NCI NIH HHS/United States
- R01 HL103532/HL/NHLBI NIH HHS/United States
- U24 CA210986/CA/NCI NIH HHS/United States
- P01 CA206978/CA/NCI NIH HHS/United States
- K08 CA248458/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
