

Compensatory sequence variation between trans-species small RNAs and their target sites
- PMID: 31845648
- PMCID: PMC6917502
- DOI: 10.7554/eLife.49750
Compensatory sequence variation between trans-species small RNAs and their target sites
Abstract
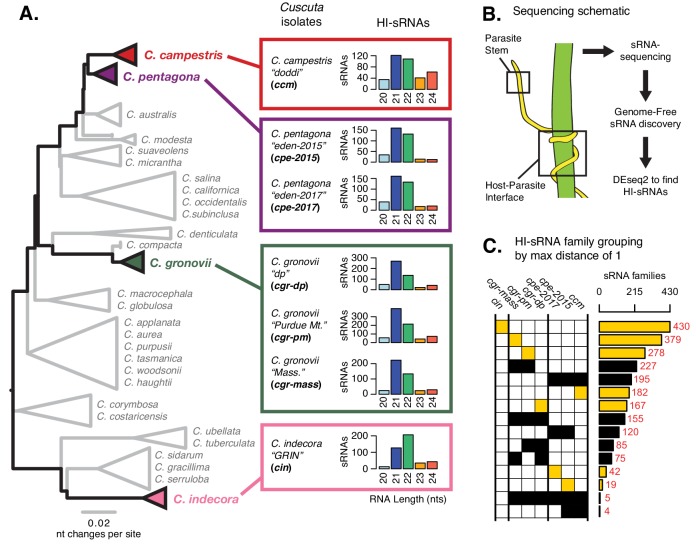
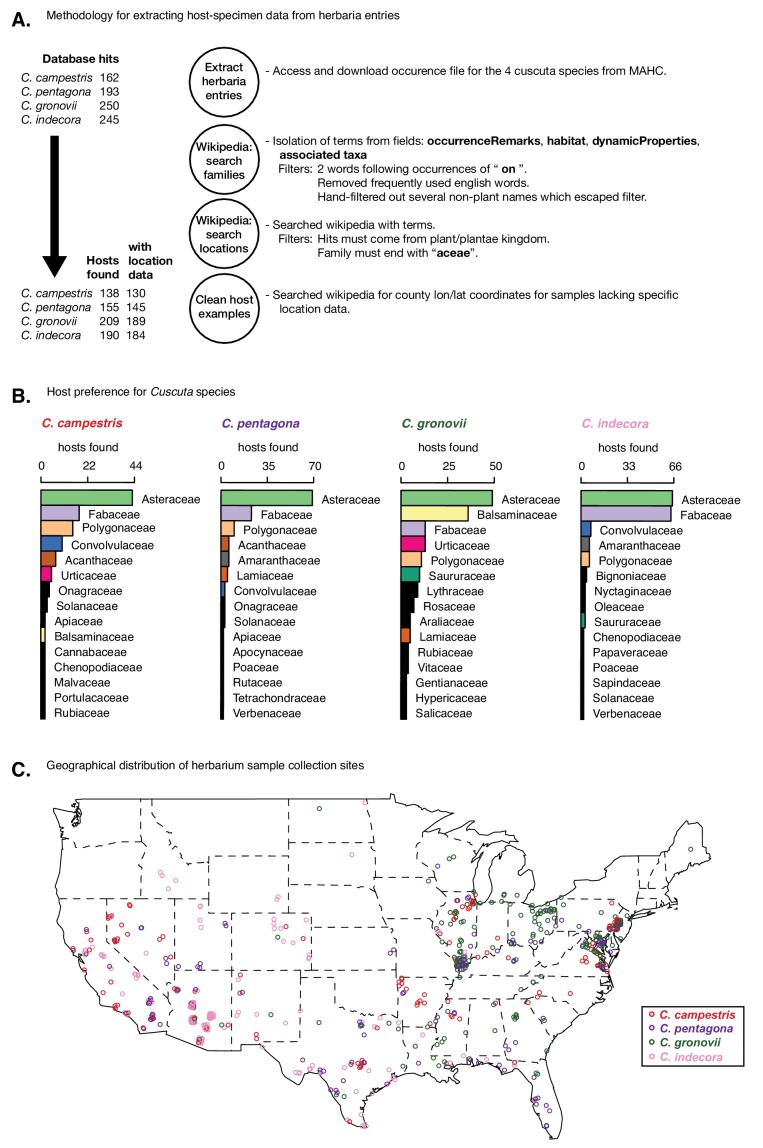
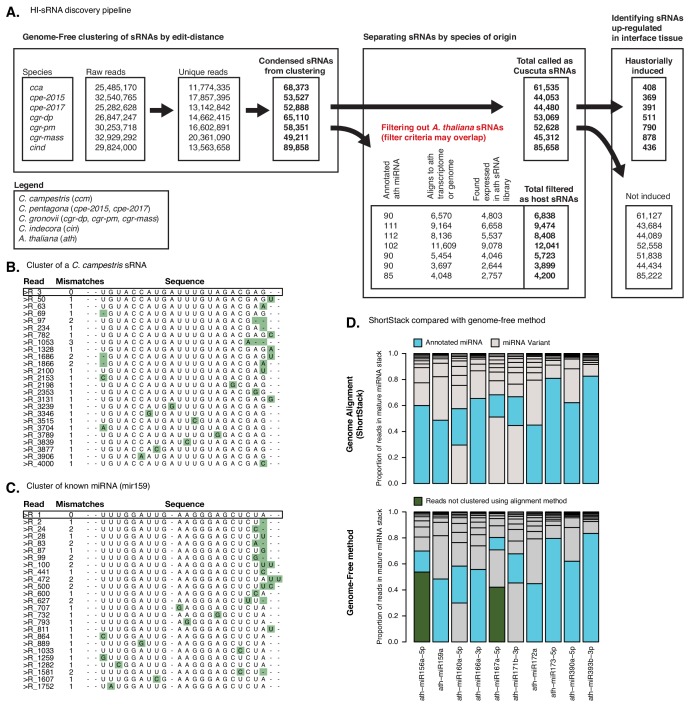
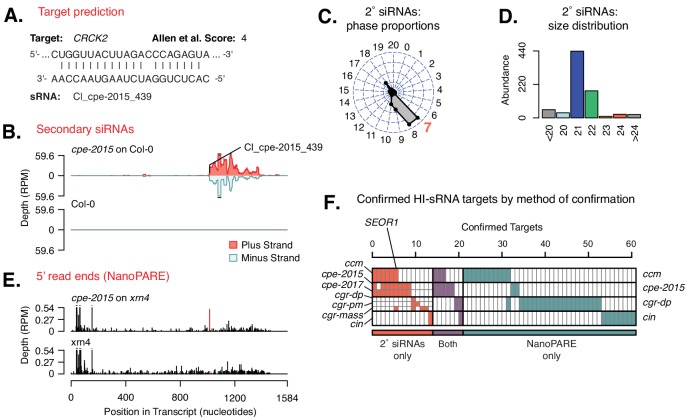
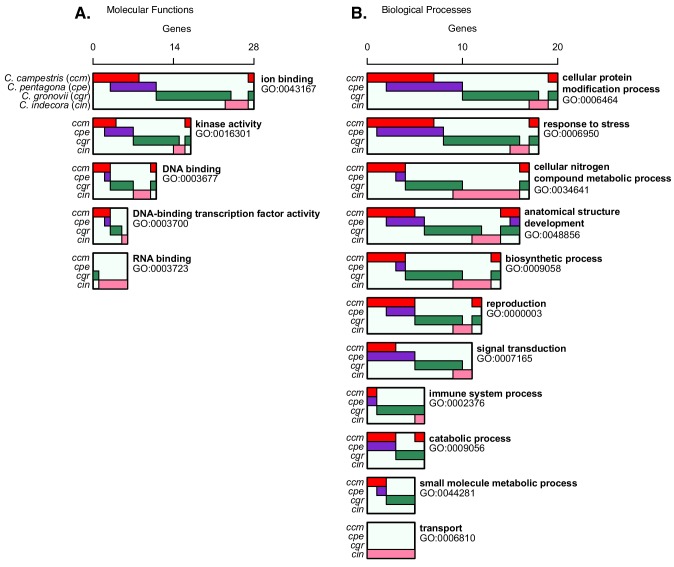
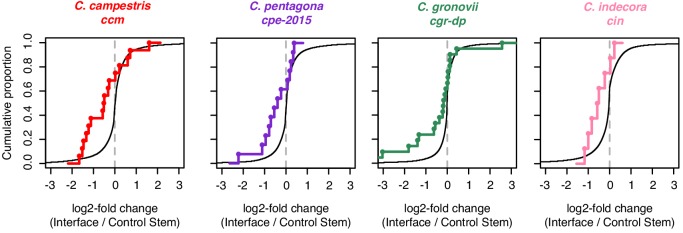
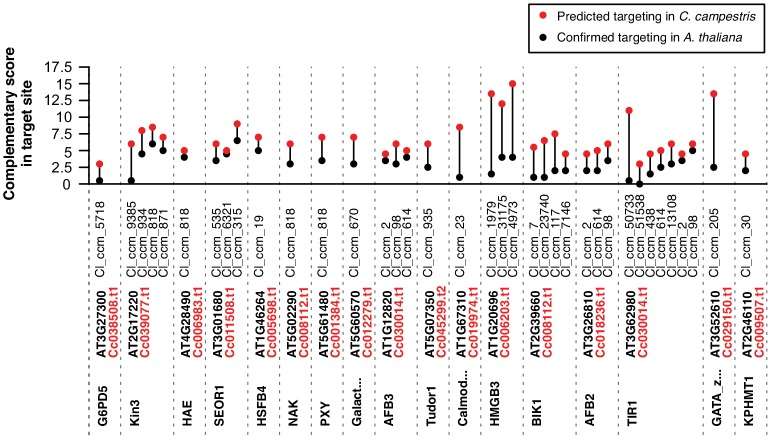
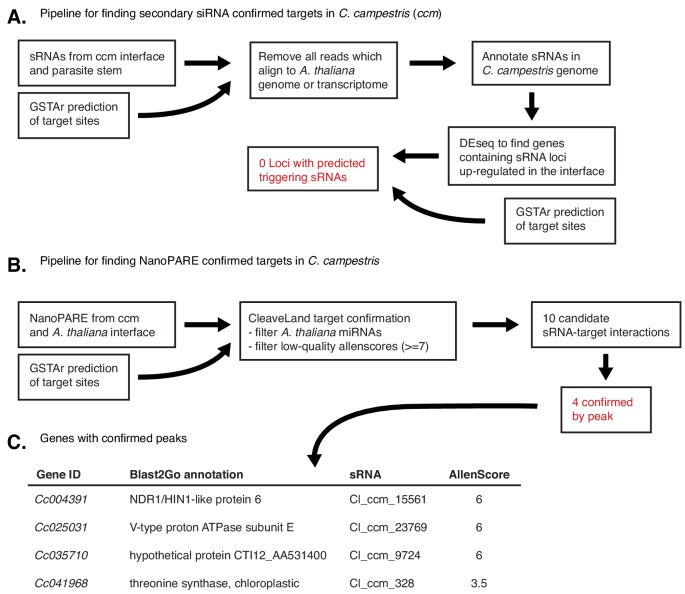
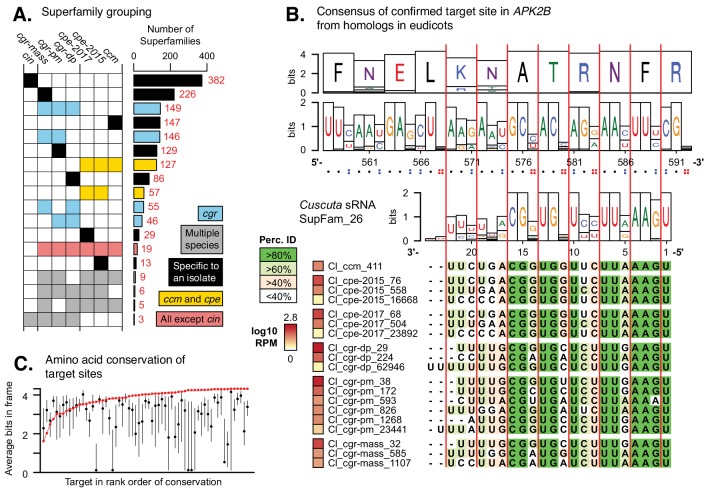
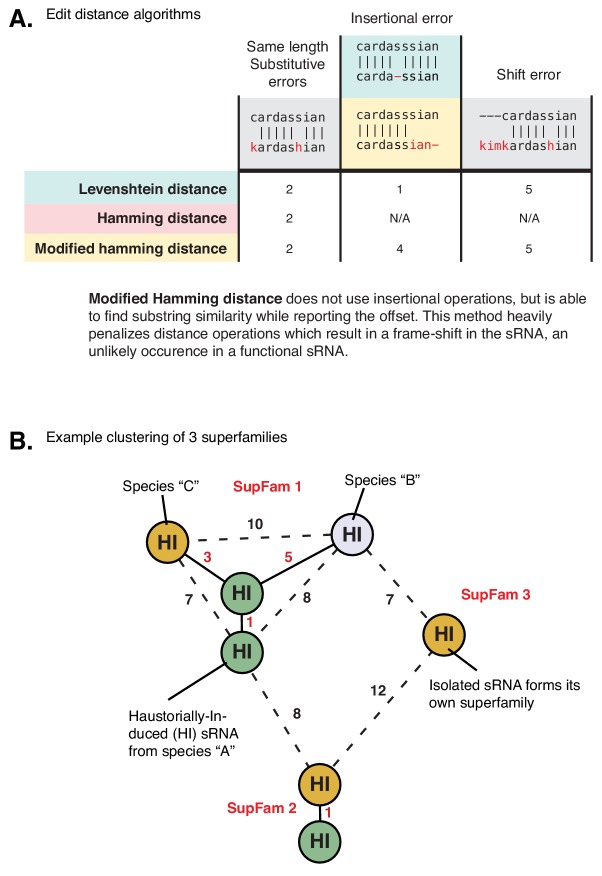
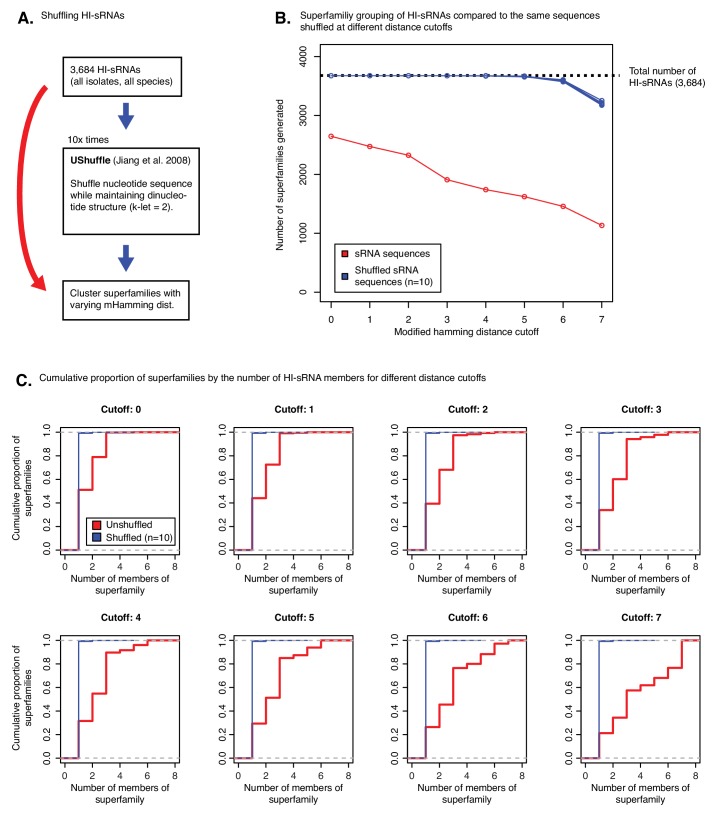
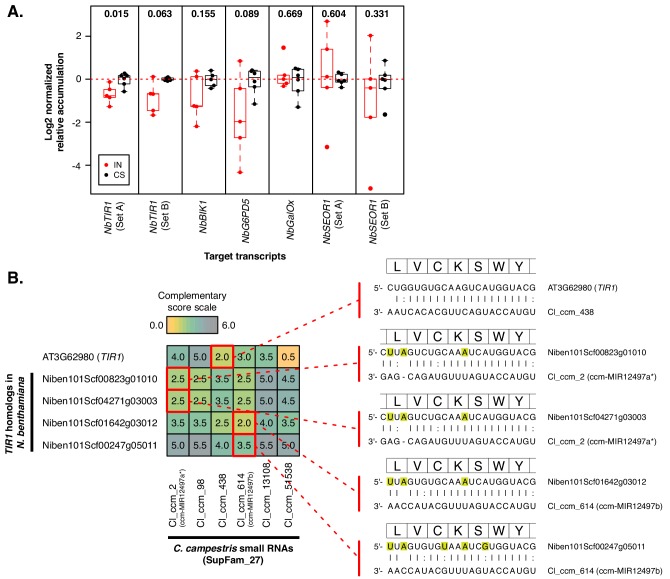
Trans-species small regulatory RNAs (sRNAs) are delivered to host plants from diverse pathogens and parasites and can target host mRNAs. How trans-species sRNAs can be effective on diverse hosts has been unclear. Multiple species of the parasitic plant Cuscuta produce trans-species sRNAs that collectively target many host mRNAs. Confirmed target sites are nearly always in highly conserved, protein-coding regions of host mRNAs. Cuscuta trans-species sRNAs can be grouped into superfamilies that have variation in a three-nucleotide period. These variants compensate for synonymous-site variation in host mRNAs. By targeting host mRNAs at highly conserved protein-coding sites, and simultaneously expressing multiple variants to cover synonymous-site variation, Cuscuta trans-species sRNAs may be able to successfully target multiple homologous mRNAs from diverse hosts.
Keywords: A. thaliana; Cuscuta; microRNA; plant biology; siRNA.
© 2019, Johnson et al.
Conflict of interest statement
NJ, Cd, MA No competing interests declared
Figures

References
-
- Axtell M. GitHub; 2014. https://github.com/MikeAxtell/GSTAr
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
