

Modeling aspects of the language of life through transfer-learning protein sequences
- PMID: 31847804
- PMCID: PMC6918593
- DOI: 10.1186/s12859-019-3220-8
Modeling aspects of the language of life through transfer-learning protein sequences
Abstract
Background: Predicting protein function and structure from sequence is one important challenge for computational biology. For 26 years, most state-of-the-art approaches combined machine learning and evolutionary information. However, for some applications retrieving related proteins is becoming too time-consuming. Additionally, evolutionary information is less powerful for small families, e.g. for proteins from the Dark Proteome. Both these problems are addressed by the new methodology introduced here.
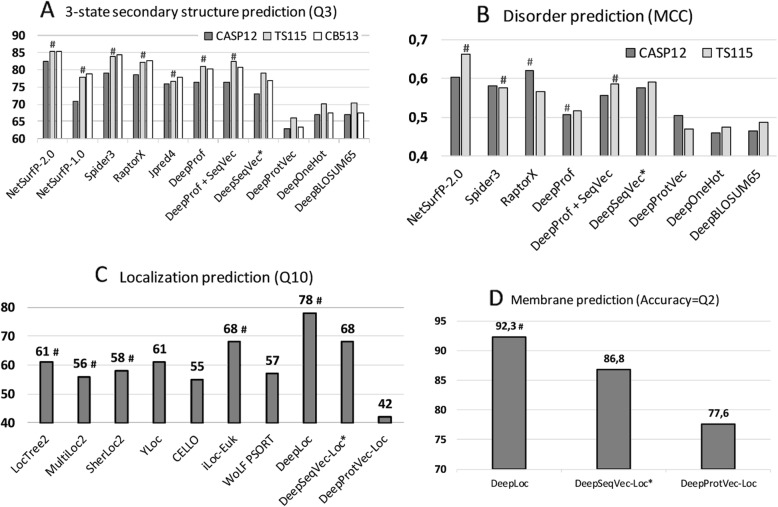
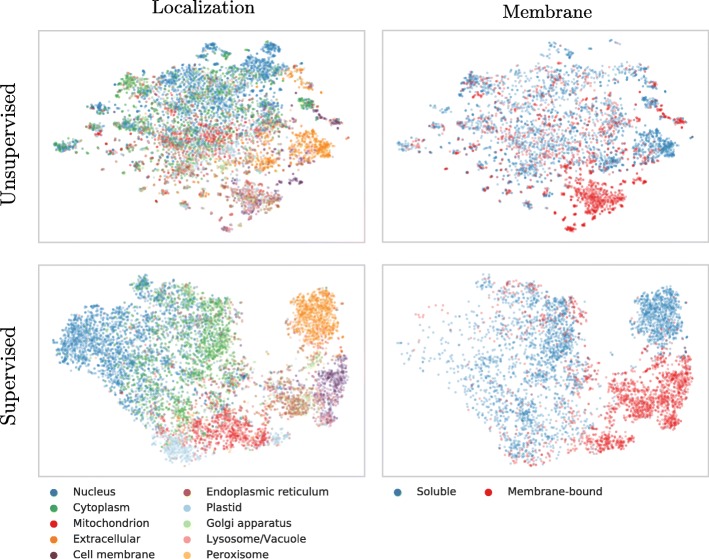
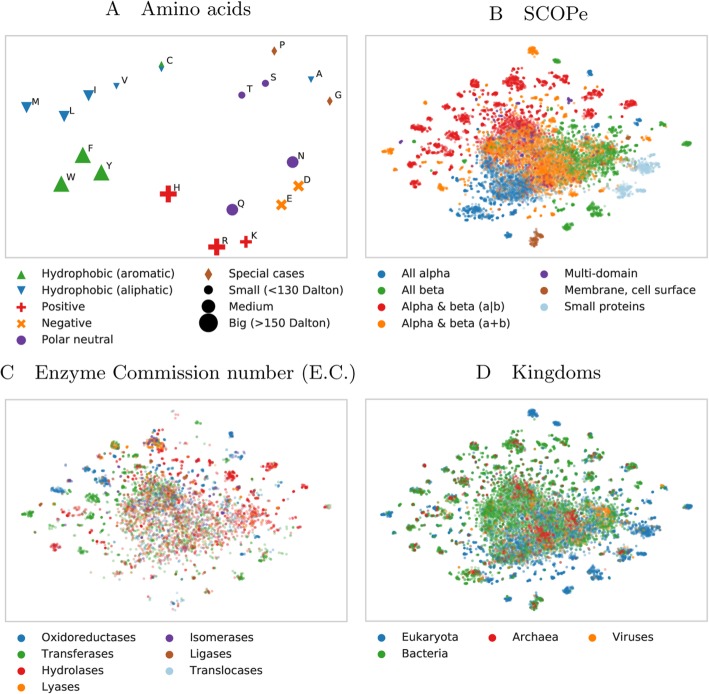
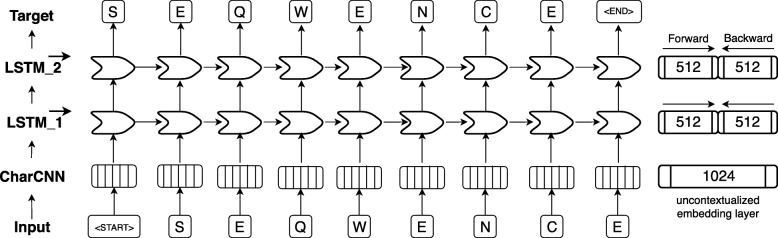
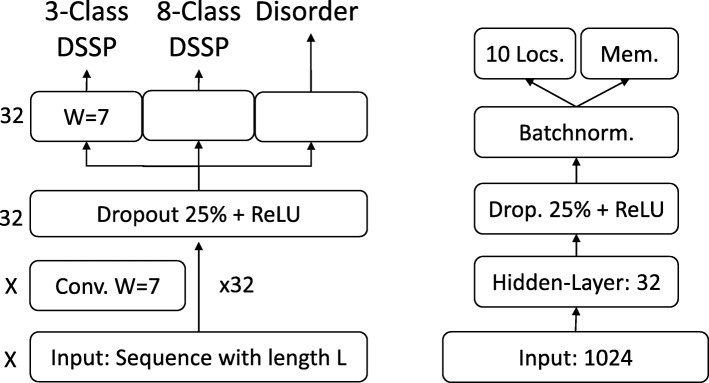
Results: We introduced a novel way to represent protein sequences as continuous vectors (embeddings) by using the language model ELMo taken from natural language processing. By modeling protein sequences, ELMo effectively captured the biophysical properties of the language of life from unlabeled big data (UniRef50). We refer to these new embeddings as SeqVec (Sequence-to-Vector) and demonstrate their effectiveness by training simple neural networks for two different tasks. At the per-residue level, secondary structure (Q3 = 79% ± 1, Q8 = 68% ± 1) and regions with intrinsic disorder (MCC = 0.59 ± 0.03) were predicted significantly better than through one-hot encoding or through Word2vec-like approaches. At the per-protein level, subcellular localization was predicted in ten classes (Q10 = 68% ± 1) and membrane-bound were distinguished from water-soluble proteins (Q2 = 87% ± 1). Although SeqVec embeddings generated the best predictions from single sequences, no solution improved over the best existing method using evolutionary information. Nevertheless, our approach improved over some popular methods using evolutionary information and for some proteins even did beat the best. Thus, they prove to condense the underlying principles of protein sequences. Overall, the important novelty is speed: where the lightning-fast HHblits needed on average about two minutes to generate the evolutionary information for a target protein, SeqVec created embeddings on average in 0.03 s. As this speed-up is independent of the size of growing sequence databases, SeqVec provides a highly scalable approach for the analysis of big data in proteomics, i.e. microbiome or metaproteome analysis.
Conclusion: Transfer-learning succeeded to extract information from unlabeled sequence databases relevant for various protein prediction tasks. SeqVec modeled the language of life, namely the principles underlying protein sequences better than any features suggested by textbooks and prediction methods. The exception is evolutionary information, however, that information is not available on the level of a single sequence.
Keywords: Deep Learning; Language Modeling; Localization prediction; Machine Learning; Secondary structure prediction; Sequence Embedding; Transfer Learning.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
