

Engineering strategies to overcome the current roadblocks in CAR T cell therapy
- PMID: 31848460
- PMCID: PMC7223338
- DOI: 10.1038/s41571-019-0297-y
Engineering strategies to overcome the current roadblocks in CAR T cell therapy
Abstract
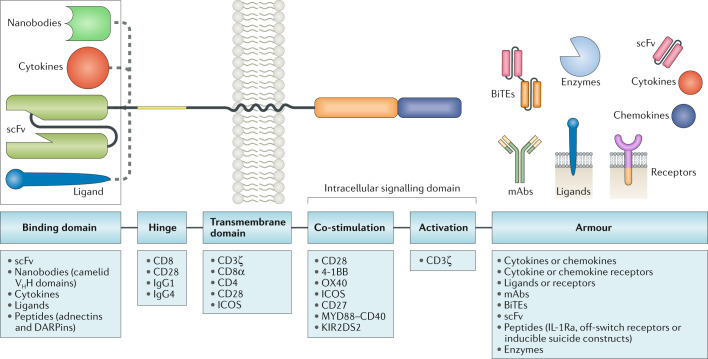
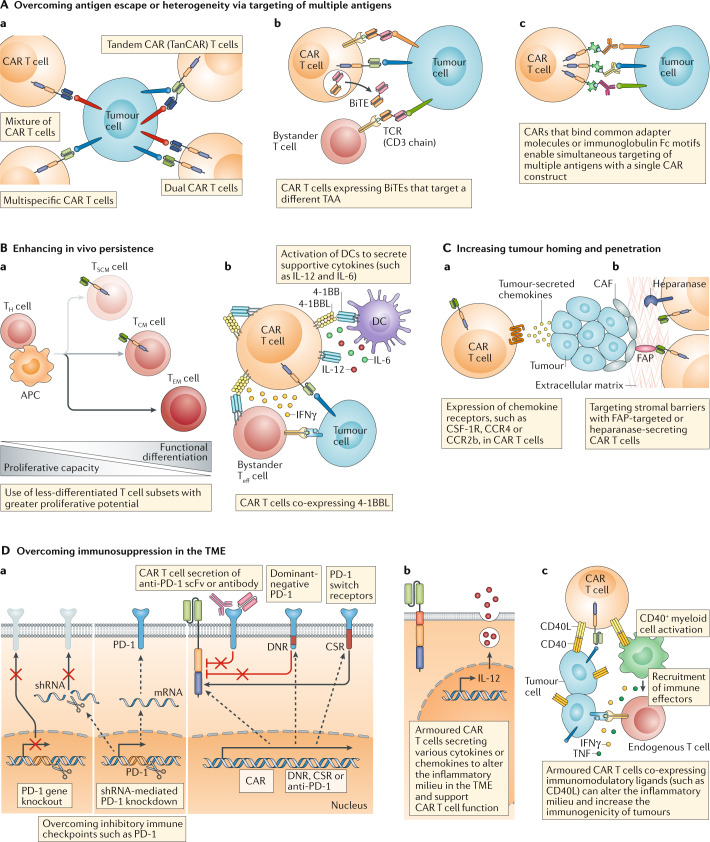
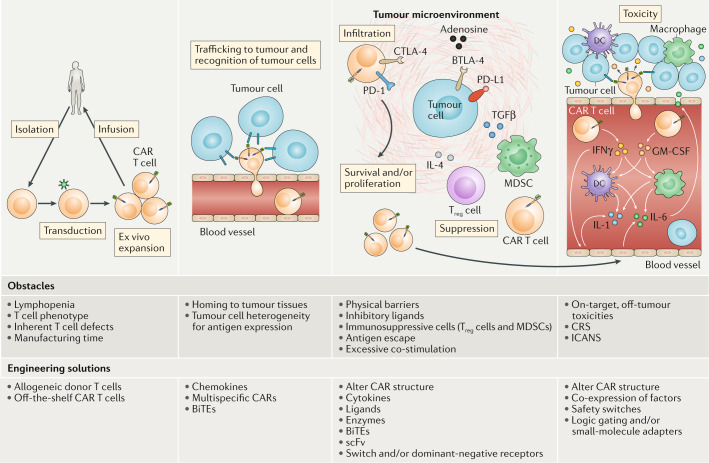
T cells genetically engineered to express chimeric antigen receptors (CARs) have proven - and impressive - therapeutic activity in patients with certain subtypes of B cell leukaemia or lymphoma, with promising efficacy also demonstrated in patients with multiple myeloma. Nevertheless, various barriers restrict the efficacy and/or prevent the widespread use of CAR T cell therapies in these patients as well as in those with other cancers, particularly solid tumours. Key challenges relating to CAR T cells include severe toxicities, restricted trafficking to, infiltration into and activation within tumours, suboptimal persistence in vivo, antigen escape and heterogeneity, and manufacturing issues. The evolution of CAR designs beyond the conventional structures will be necessary to address these limitations and to expand the use of CAR T cells to a wider range of malignancies. Investigators are addressing the current obstacles with a wide range of engineering strategies in order to improve the safety, efficacy and applicability of this therapeutic modality. In this Review, we discuss the innovative designs of novel CAR T cell products that are being developed to increase and expand the clinical benefits of these treatments in patients with diverse cancers.
Conflict of interest statement
R.J.B. receives royalties and grant support from JUNO Therapeutics and is a consultant for JUNO Therapeutics/Celgene. The other authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
