

Lipid metabolic pathways converge in motor neuron degenerative diseases
- PMID: 31848577
- PMCID: PMC7174042
- DOI: 10.1093/brain/awz382
Lipid metabolic pathways converge in motor neuron degenerative diseases
Abstract
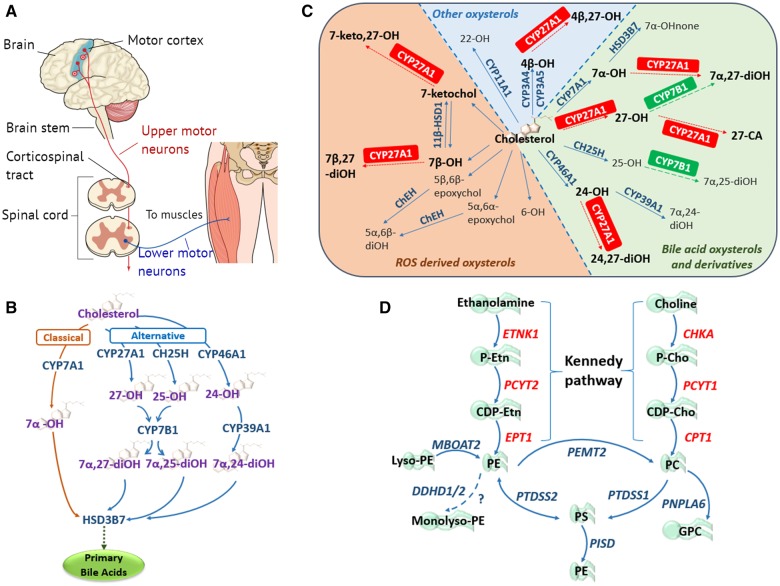
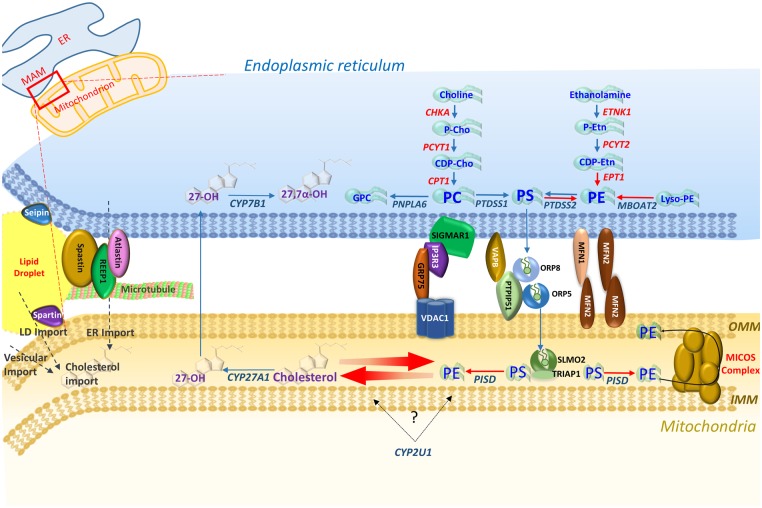
Motor neuron diseases (MNDs) encompass an extensive and heterogeneous group of upper and/or lower motor neuron degenerative disorders, in which the particular clinical outcomes stem from the specific neuronal component involved in each condition. While mutations in a large number of molecules associated with lipid metabolism are known to be implicated in MNDs, there remains a lack of clarity regarding the key functional pathways involved, and their inter-relationships. This review highlights evidence that defines defects within two specific lipid (cholesterol/oxysterol and phosphatidylethanolamine) biosynthetic cascades as being centrally involved in MND, particularly hereditary spastic paraplegia. We also identify how other MND-associated molecules may impact these cascades, in particular through impaired organellar interfacing, to propose 'subcellular lipidome imbalance' as a likely common pathomolecular theme in MND. Further exploration of this mechanism has the potential to identify new therapeutic targets and management strategies for modulation of disease progression in hereditary spastic paraplegias and other MNDs.
Keywords: HSP; MND; cholesterol; lipidome imbalance; mitochondria.
© The Author(s) (2019). Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Comment in
-
Defective phosphatidylethanolamine biosynthesis leads to a broad ataxia-spasticity spectrum.Brain. 2021 Apr 12;144(3):e30. doi: 10.1093/brain/awaa442. Brain. 2021. PMID: 33454747 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
