Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type
- PMID: 31849330
- PMCID: PMC6918713
- DOI: 10.1186/s13059-019-1892-z
Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type
Abstract
Background: The lifelong accumulation of somatic mutations underlies age-related phenotypes and cancer. Mutagenic forces are thought to shape the genome of aging cells in a tissue-specific way. Whole genome analyses of somatic mutation patterns, based on both types and genomic distribution of variants, can shed light on specific processes active in different human tissues and their effect on the transition to cancer.
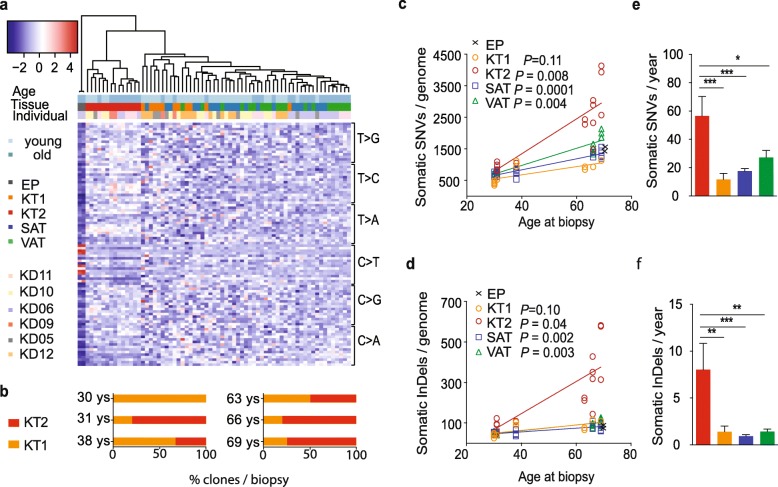
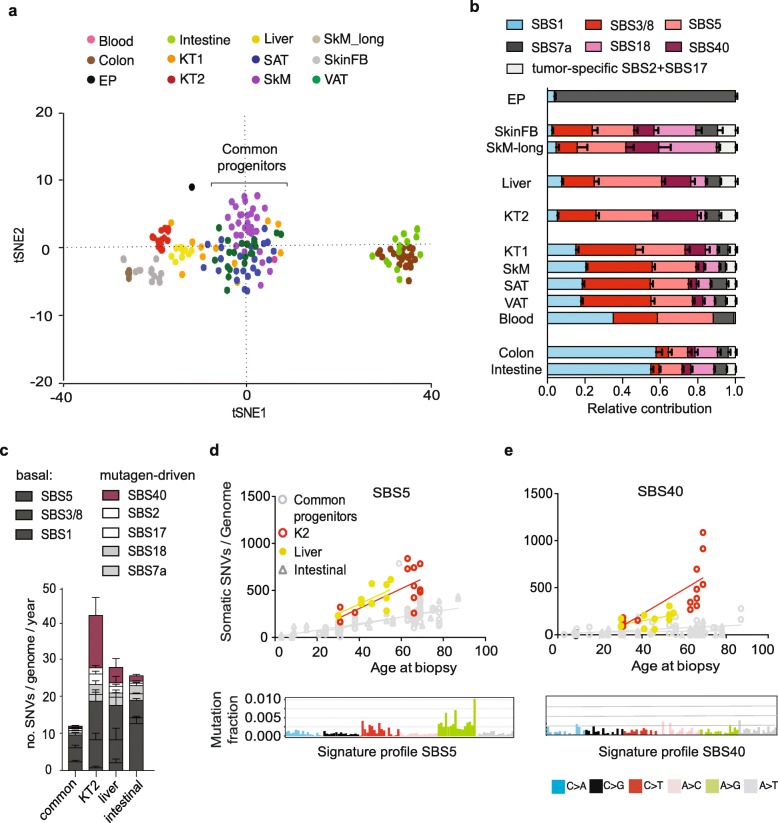
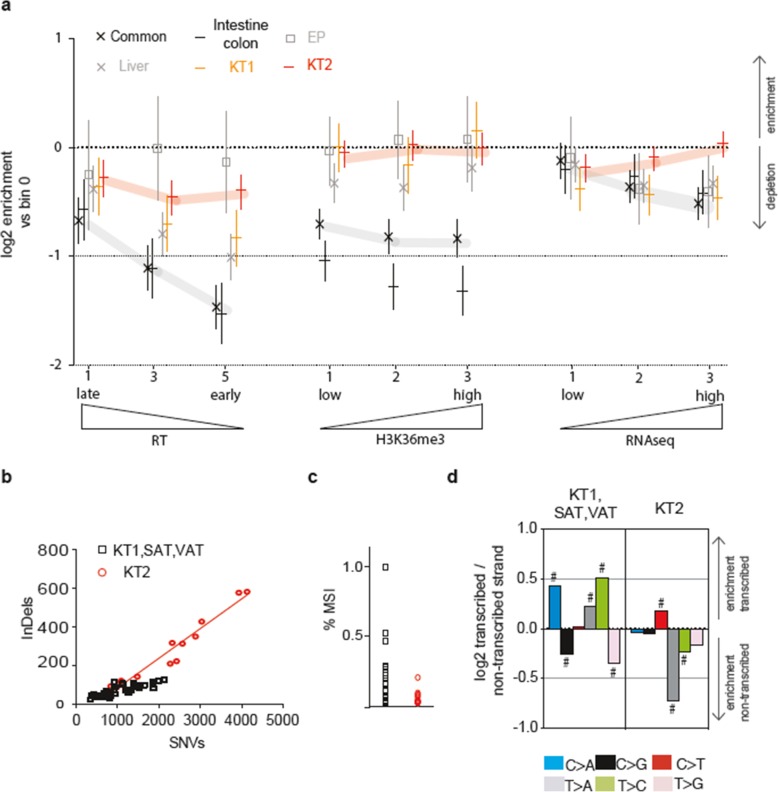
Results: To analyze somatic mutation patterns, we compile a comprehensive genetic atlas of somatic mutations in healthy human cells. High-confidence variants are obtained from newly generated and publicly available whole genome DNA sequencing data from single non-cancer cells, clonally expanded in vitro. To enable a well-controlled comparison of different cell types, we obtain single genome data (92% mean coverage) from multi-organ biopsies from the same donors. These data show multiple cell types that are protected from mutagens and display a stereotyped mutation profile, despite their origin from different tissues. Conversely, the same tissue harbors cells with distinct mutation profiles associated to different differentiation states. Analyses of mutation rate in the coding and non-coding portions of the genome identify a cell type bearing a unique mutation pattern characterized by mutation enrichment in active chromatin, regulatory, and transcribed regions.
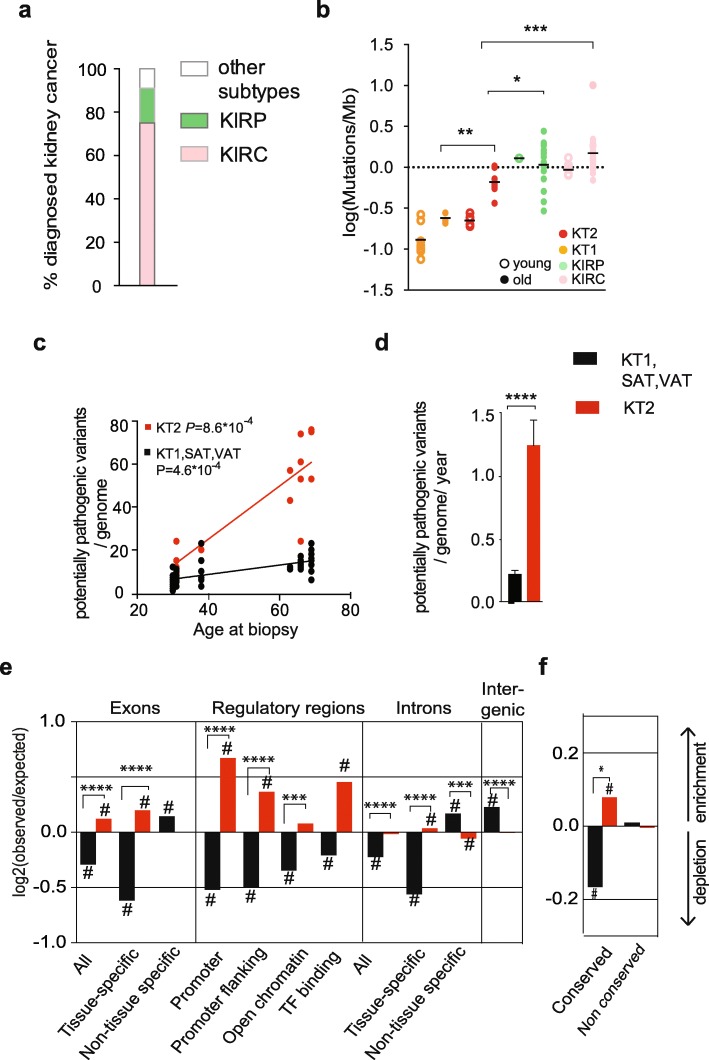
Conclusions: Our analysis of normal cells from healthy donors identifies a somatic mutation landscape that enhances the risk of tumor transformation in a specific cell population from the kidney proximal tubule. This unique pattern is characterized by high rate of mutation accumulation during adult life and specific targeting of expressed genes and regulatory regions.
Keywords: Aging; Kidney cancer; Proximal tubule; Somatic mutations; kidney progenitors.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures