

Toward the Accuracy and Speed of Protein Side-Chain Packing: A Systematic Study on Rotamer Libraries
- PMID: 31851497
- PMCID: PMC7938712
- DOI: 10.1021/acs.jcim.9b00812
Toward the Accuracy and Speed of Protein Side-Chain Packing: A Systematic Study on Rotamer Libraries
Abstract
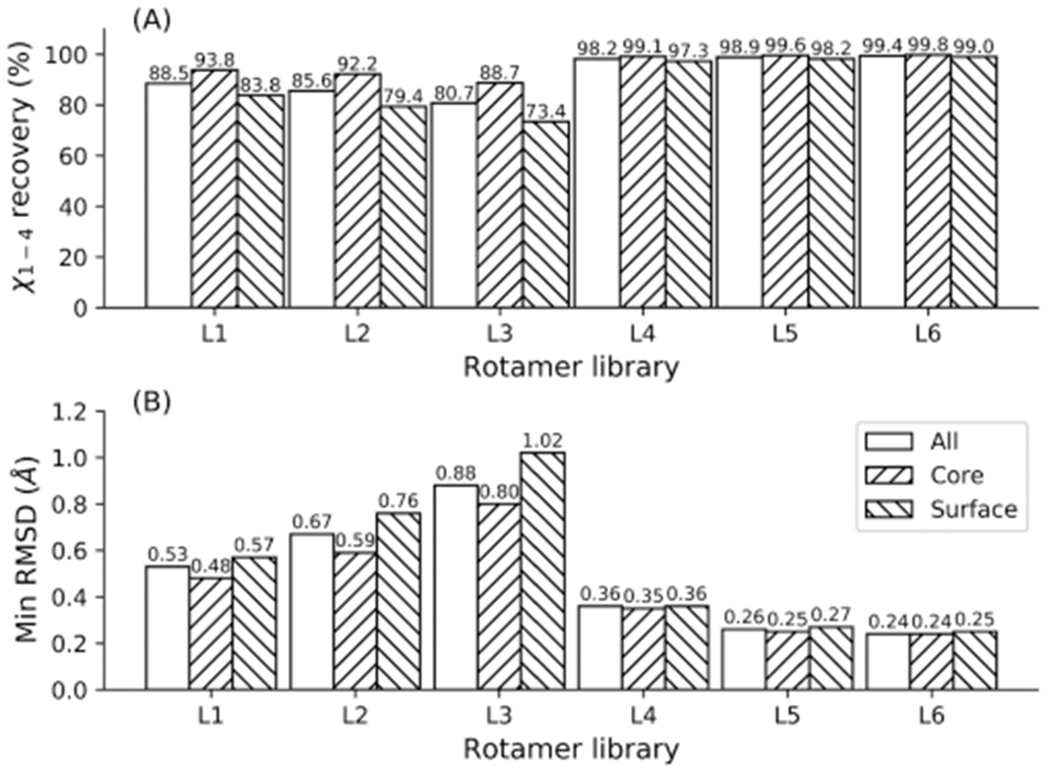
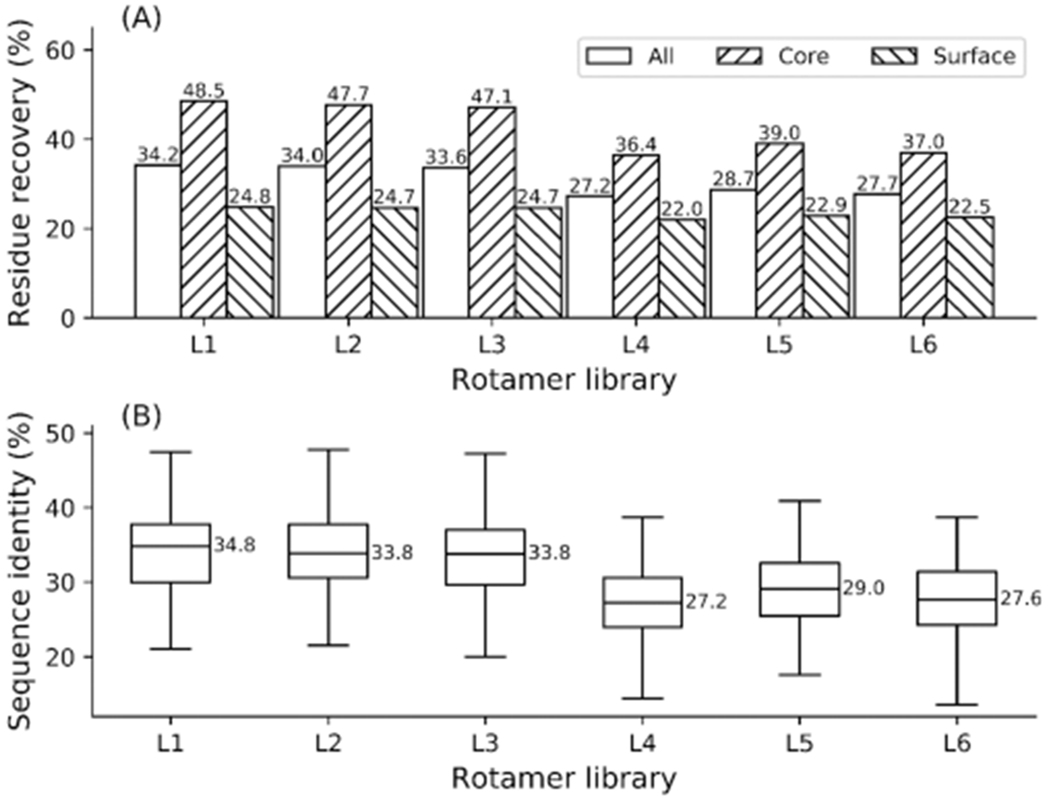
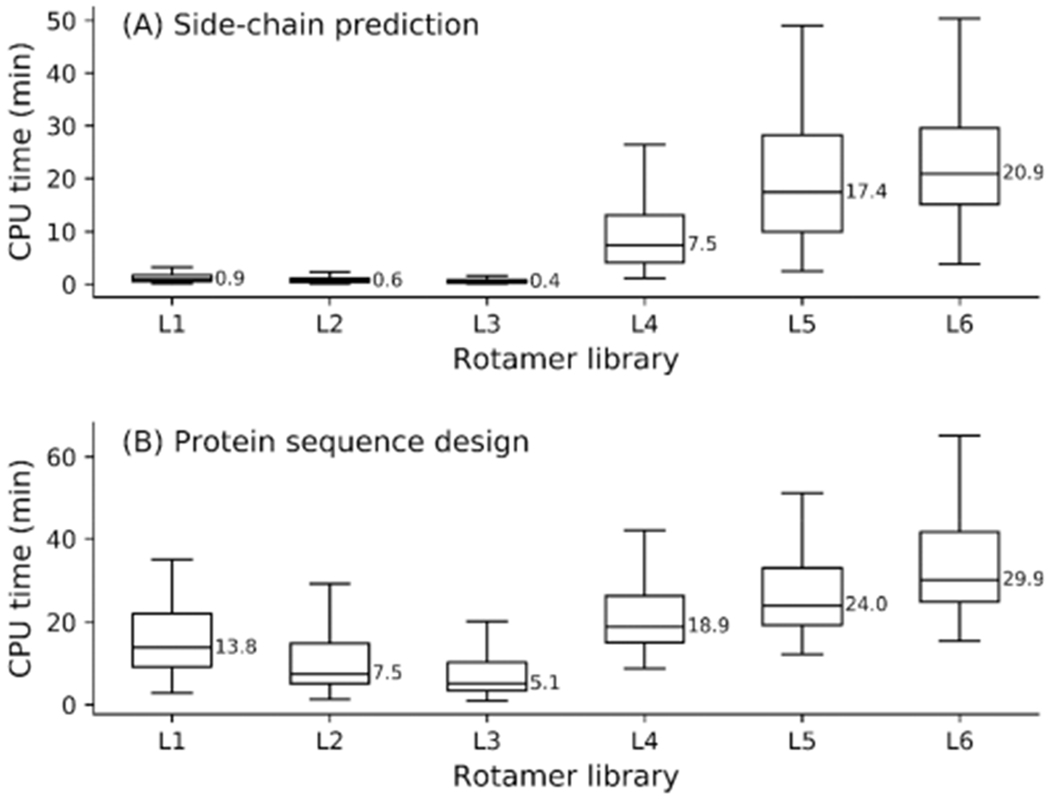
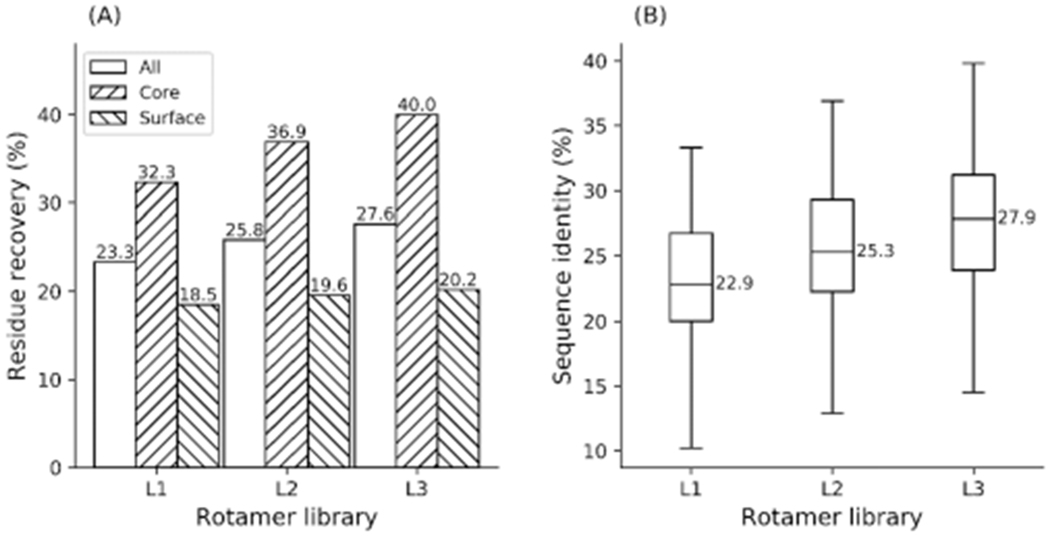
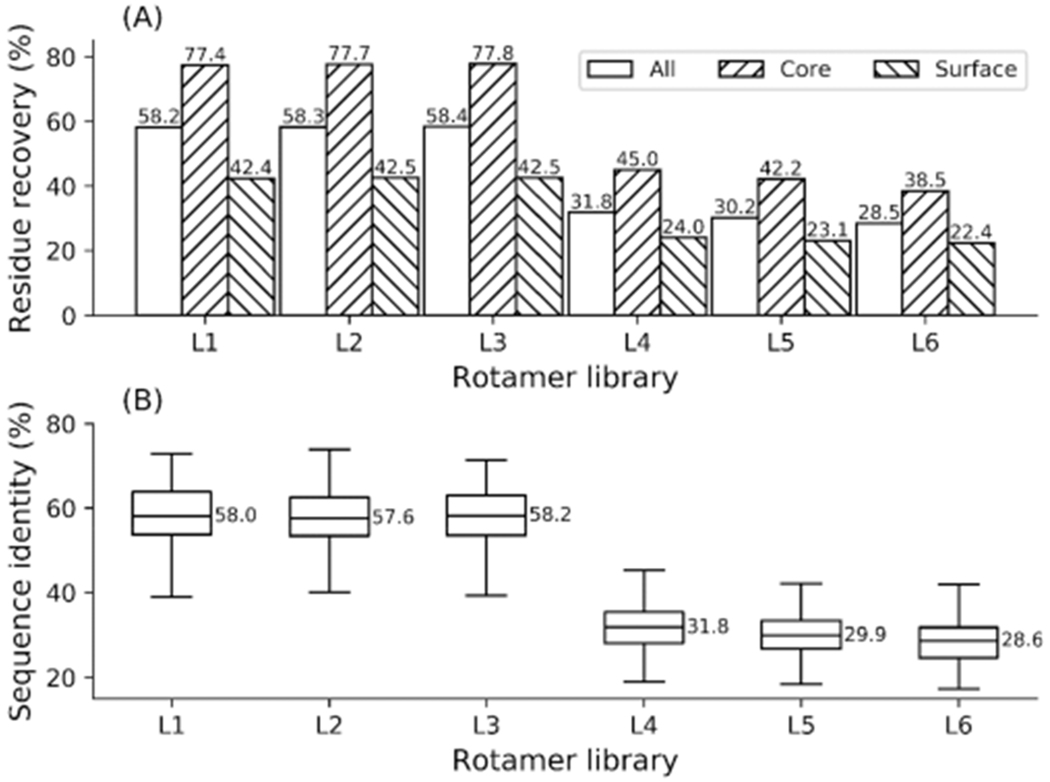
Protein rotamers refer to the conformational isomers taken by the side-chains of amino acids to accommodate specific structural folding environments. Since accurate modeling of atomic interactions is difficult, rotamer information collected from experimentally solved protein structures is often used to guide side-chain packing in protein folding and sequence design studies. Many rotamer libraries have been built in the literature but there is little quantitative guidance on which libraries should be chosen for different structural modeling studies. Here, we performed a comparative study of six widely used rotamer libraries and systematically examined their suitability for protein folding and sequence design in four aspects: (1) side-chain match accuracy, (2) side-chain conformation prediction, (3) de novo protein sequence design, and (4) computational time cost. We demonstrated that, compared to the backbone-dependent rotamer libraries (BBDRLs), the backbone-independent rotamer libraries (BBIRLs) generated conformations that more closely matched the native conformations due to the larger number of rotamers in the local rotamer search spaces. However, more practically, using an optimized physical energy function incorporated into a simulated annealing Monte Carlo searching scheme, we showed that utilization of the BBDRLs could result in higher accuracies in side-chain prediction and higher sequence recapitulation rates in protein design experiments. Detailed data analyses showed that the major advantage of BBDRLs lies in the energy term derived from the rotamer probabilities that are associated with the individual backbone torsion angle subspaces. This term is important for distinguishing between amino acid identities as well as the rotamer conformations of an amino acid. Meanwhile, the backbone torsion angle subspace-specific rotamer search drastically speeds up the searching time, despite the significantly larger number of total rotamers in the BBDRLs. These results should provide important guidance for the development and selection of rotamer libraries for practical protein design and structure prediction studies.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
A protein-dependent side-chain rotamer library.BMC Bioinformatics. 2011 Dec 14;12 Suppl 14(Suppl 14):S10. doi: 10.1186/1471-2105-12-S14-S10. BMC Bioinformatics. 2011. PMID: 22373394 Free PMC article.
-
Rotamer libraries and probabilities of transition between rotamers for the side chains in protein-protein binding.Proteins. 2012 Aug;80(8):2089-98. doi: 10.1002/prot.24103. Epub 2012 Jun 12. Proteins. 2012. PMID: 22544766 Free PMC article.
-
A backbone-dependent rotamer library with high (ϕ, ψ) coverage using metadynamics simulations.Protein Sci. 2022 Dec;31(12):e4491. doi: 10.1002/pro.4491. Protein Sci. 2022. PMID: 36327064 Free PMC article.
-
Rotamer Dynamics: Analysis of Rotamers in Molecular Dynamics Simulations of Proteins.Biophys J. 2019 Jun 4;116(11):2062-2072. doi: 10.1016/j.bpj.2019.04.017. Epub 2019 Apr 22. Biophys J. 2019. PMID: 31084902 Free PMC article. Review.
-
Rotamer libraries in the 21st century.Curr Opin Struct Biol. 2002 Aug;12(4):431-40. doi: 10.1016/s0959-440x(02)00344-5. Curr Opin Struct Biol. 2002. PMID: 12163064 Review.
Cited by
-
Exploiting Sequence-Dependent Rotamer Information in Global Optimization of Proteins.J Phys Chem B. 2022 Oct 27;126(42):8381-8390. doi: 10.1021/acs.jpcb.2c04647. Epub 2022 Oct 18. J Phys Chem B. 2022. PMID: 36257022 Free PMC article.
-
GeoPacker: A novel deep learning framework for protein side-chain modeling.Protein Sci. 2022 Dec;31(12):e4484. doi: 10.1002/pro.4484. Protein Sci. 2022. PMID: 36309961 Free PMC article.
-
Invariant point message passing for protein side chain packing.Proteins. 2024 Oct;92(10):1220-1233. doi: 10.1002/prot.26705. Epub 2024 May 24. Proteins. 2024. PMID: 38790143
-
Decoding CRISPR-Cas PAM recognition with UniDesign.Brief Bioinform. 2023 May 19;24(3):bbad133. doi: 10.1093/bib/bbad133. Brief Bioinform. 2023. PMID: 37078688 Free PMC article.
-
Identifying zoonotic origin of SARS-CoV-2 by modeling the binding affinity between Spike receptor-binding domain and host ACE2.bioRxiv [Preprint]. 2020 Sep 11:2020.09.11.293449. doi: 10.1101/2020.09.11.293449. bioRxiv. 2020. Update in: J Proteome Res. 2020 Dec 4;19(12):4844-4856. doi: 10.1021/acs.jproteome.0c00717. PMID: 32935105 Free PMC article. Updated. Preprint.
References
-
- Colbes J; Corona RI; Lezcano C; Rodriguez D; Brizuela CA Protein side-chain packing problem: is there still room for improvement? Briefings Bioinf. 2016, 18, 1033–1043. - PubMed
-
- Huang X; Yang J; Zhu Y A solvated ligand rotamer approach and its application in computational protein design. J. Mol. Model 2013, 19, 1355–1367. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
