

The impact of oncogenic RAS on redox balance and implications for cancer development
- PMID: 31852884
- PMCID: PMC6920345
- DOI: 10.1038/s41419-019-2192-y
The impact of oncogenic RAS on redox balance and implications for cancer development
Abstract
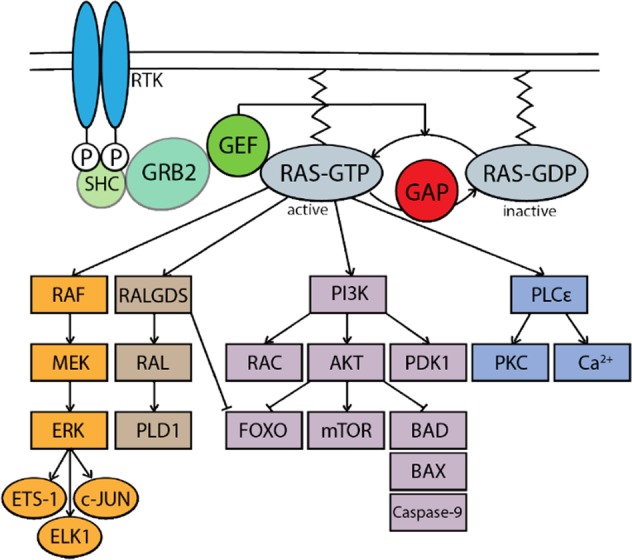
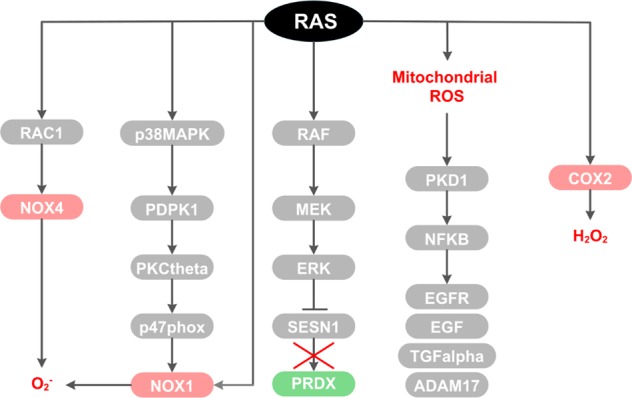
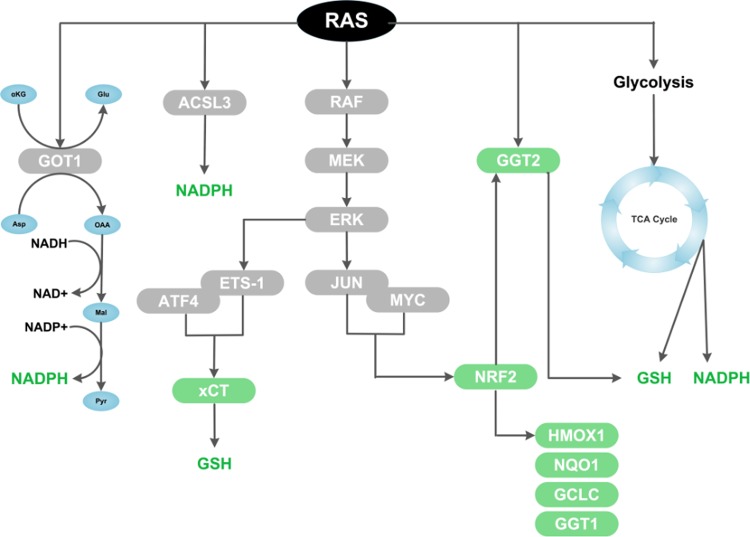
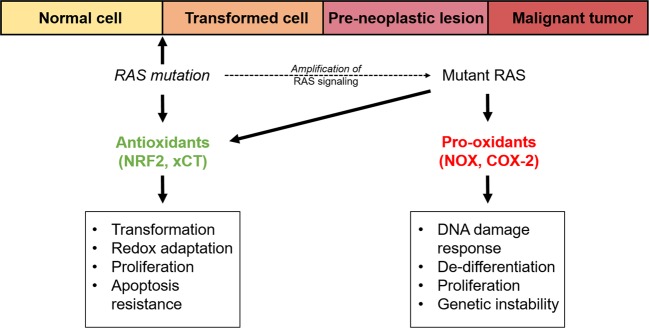
The RAS family of proto-oncogenes comprises HRAS, KRAS, and NRAS, which are among the most mutated genes in human cancers. The RAS family genes encode small GTPases that coordinate key signaling pathways in response to growth factors. Mutations in RAS result in a constitutively active form of the protein that supports cellular transformation and tumorigenesis. The mechanisms of oncogenic RAS-mediated transformation encompass uncontrolled proliferation and inhibition of cell death through overactivation of the RAF-MEK-ERK and the PI3K-AKT pathways, respectively. In addition, the control of redox balance by RAS has also been proposed to play a role in its oncogenic properties. However, the exact role of redox balance in mediating mutant RAS transformation is still under debate. Here, we present, on one hand, the involvement of pro-oxidant components in oncogenic RAS transformation, such as NADPH oxidases and mitochondrial reactive oxygen species, and how these promote transformation. On the other hand, we describe the contribution of antioxidant components to mutant RAS transformation, including Nrf2, glutathione biosynthesis and xCT, as well as the mechanisms by which antioxidant programs drive transformation. Finally, we aim to reconcile the seemingly opposite effects of oncogenic RAS on redox balance and discuss a model for the complementary role of both pro-oxidant and antioxidant pathways in mutant RAS-driven tumor progression.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
