

CD8+ T cell-mediated endotheliopathy is a targetable mechanism of neuro-inflammation in Susac syndrome
- PMID: 31852955
- PMCID: PMC6920411
- DOI: 10.1038/s41467-019-13593-5
CD8+ T cell-mediated endotheliopathy is a targetable mechanism of neuro-inflammation in Susac syndrome
Abstract
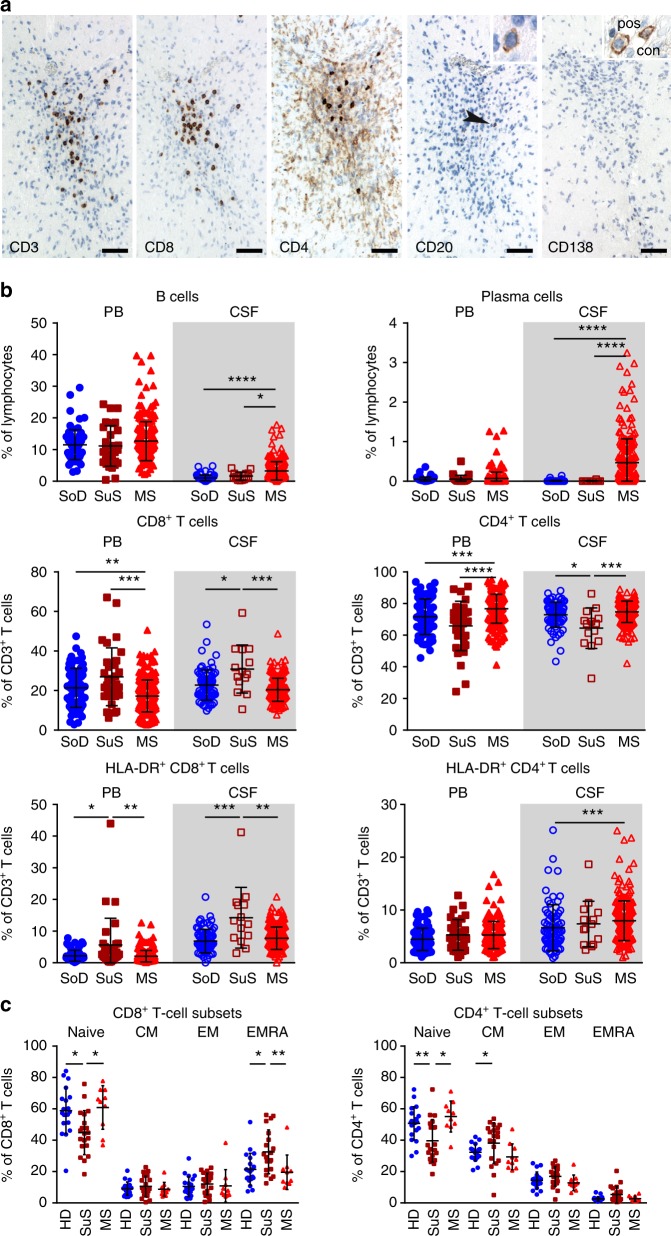
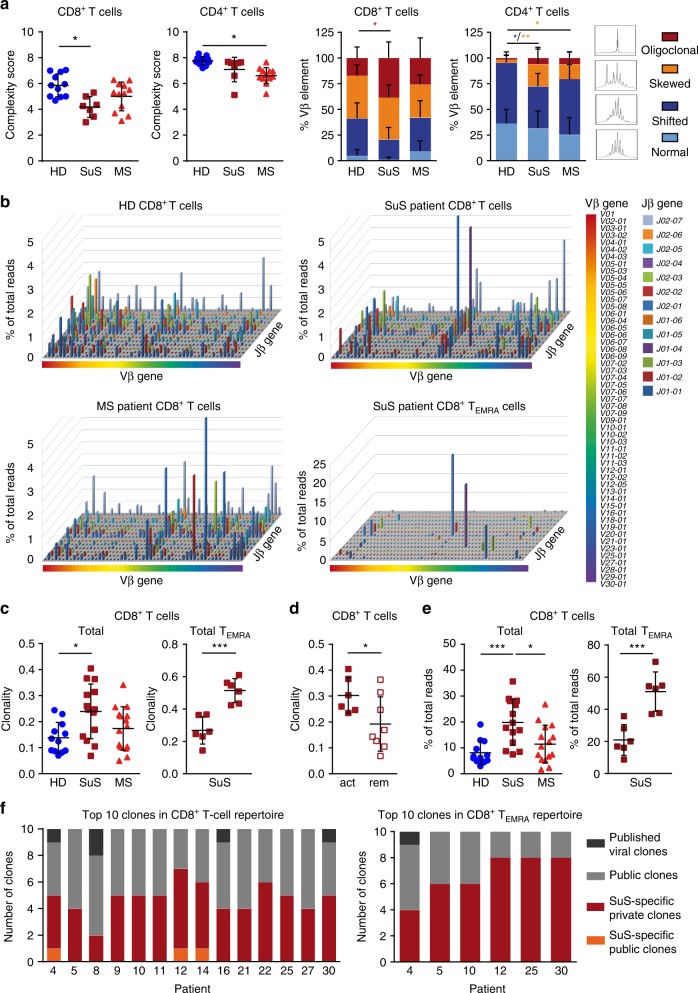
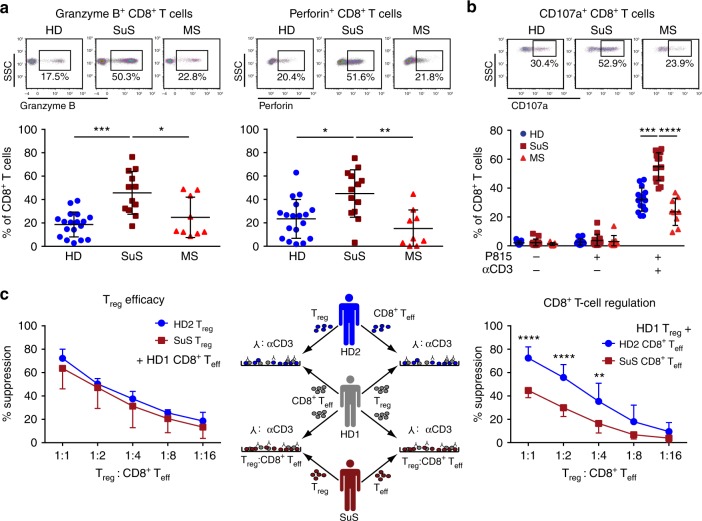
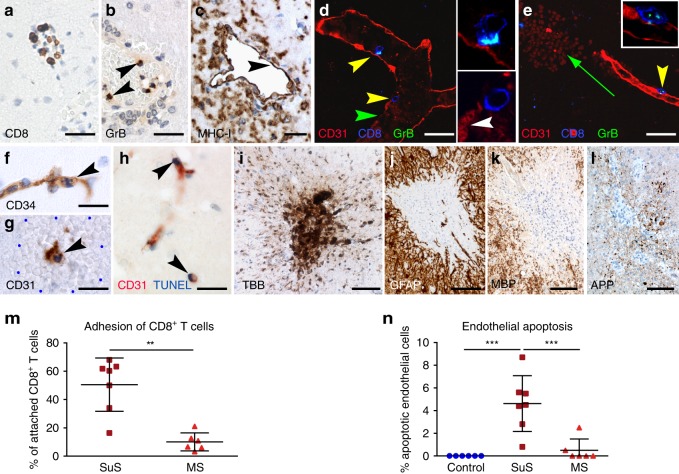
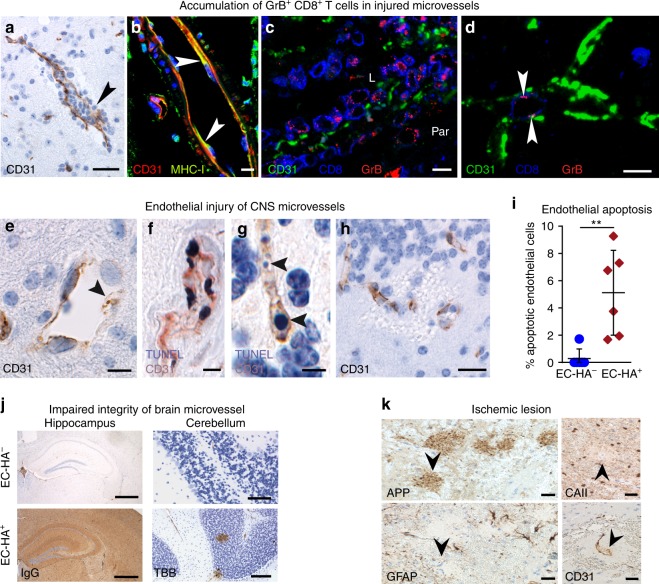
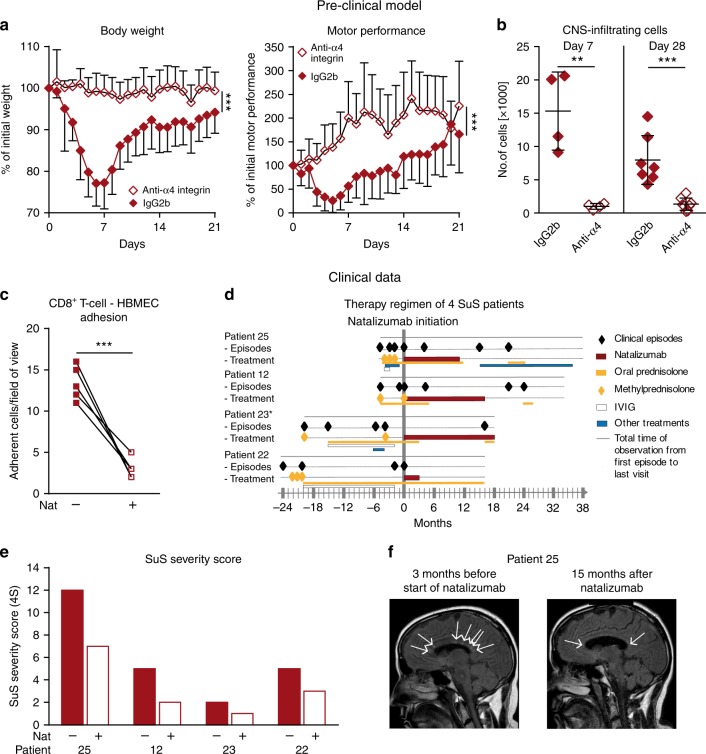
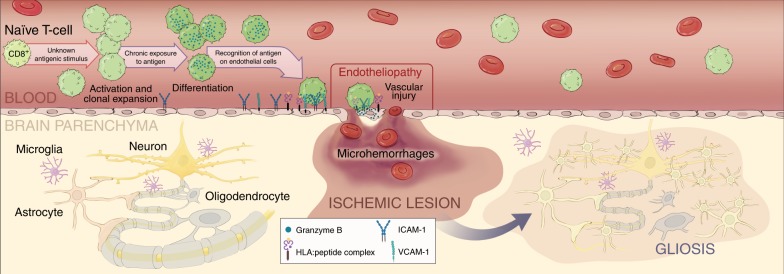
Neuroinflammation is often associated with blood-brain-barrier dysfunction, which contributes to neurological tissue damage. Here, we reveal the pathophysiology of Susac syndrome (SuS), an enigmatic neuroinflammatory disease with central nervous system (CNS) endotheliopathy. By investigating immune cells from the blood, cerebrospinal fluid, and CNS of SuS patients, we demonstrate oligoclonal expansion of terminally differentiated activated cytotoxic CD8+ T cells (CTLs). Neuropathological data derived from both SuS patients and a newly-developed transgenic mouse model recapitulating the disease indicate that CTLs adhere to CNS microvessels in distinct areas and polarize granzyme B, which most likely results in the observed endothelial cell injury and microhemorrhages. Blocking T-cell adhesion by anti-α4 integrin-intervention ameliorates the disease in the preclinical model. Similarly, disease severity decreases in four SuS patients treated with natalizumab along with other therapy. Our study identifies CD8+ T-cell-mediated endotheliopathy as a key disease mechanism in SuS and highlights therapeutic opportunities.
Conflict of interest statement
C.C.G. received speaker honoraria and travel expenses for attending meetings from Biogen, Euroimmun, Genzyme, MyLan, Novartis Pharma GmbH, and Bayer Health Care, none related to this study. Her work is funded by Biogen, Novartis, the German Ministry for Education and Research (BMBF; 01Gl1603A), the German Research Foundation (DFG, GR3946/3–1, SFB Transregio 128 A09), the European Union (Horizon2020, ReSToRE), the Interdisciplinary Center for Clinical Studies (IZKF), and the IMF. I.K. received travel expenses for attending meetings from Pfizer and CSL Behring. I.K. received speaker honoraria from Daiichi Sankyo. J.B.’s work is funded by the Austrian Science Fund (FWF: P26936-B27). A.S.-M. receives research support from Novartis. T.K. received speaker honoraria from Novartis and Excemed. Her research is supported by the DFG (SFB/Transregio 128 B07), European Leukodystrophy Association (2017-018C4A), National MS Society (RG-1801-30020), and Progressive MS Alliance (PA-1604-08492, VRAVEinMS). W.B. has received honoraria for lectures by Bayer Vital, Biogen, Merck Serono, Teva Pharma, Genzyme, Sanofi-Aventis, Novartis, Excemed, and Medday. He is a member of scientific advisory boards for Teva Pharma, Biogen, Novartis, and Genzyme. W.B. receives research support from Teva Pharma, Biogen, Genzyme, and Novartis. W.B. is supported by the Deutsche Forschungsgemeinschaft, the German Ministry for Education and Research, and by the Klaus Tschira Foundation. M.P. received speaker honoraria from Roche and Genzyme and travel/accommodation/meeting expenses from Novartis, Biogen Idec, Genzyme, and MERCK Serono. D.-A.L. received speaker honoraria from Biogen, Merck, Novartis, Roche, Teva, and Genzyme and non-personal grants from Biogen, Genzyme, Novartis, MedDay, Merck, and Roche not related to this study. J.P. is a member of scientific advisory boards from Genzyme and Novartis. He received honoraria from Biogen, Sanofi/Genzyme, and Merck Serono. M.B. received institutional support for research, speaking and/or participation in advisory boards for Biogen, Merck, Novartis, Roche, and Sanofi Genzyme. He is a consulting neurologist for RxMx/Medical Safety Systems and a research director for Sydney Neuroimaging Analysis Centre. T.A.H. has received honoraria or travel sponsorship from Bayer-Schering, Novartis, Biogen Idec, Merck-Serono, Roche, Teva, Alexion, and Sanofi-Genzyme. S.W.R. has received honoraria, travel sponsorship, research, and/or departmental support from MGANSW, MGAQLD, MAA, Lambert Initiative, Beeren foundation, anonymous donors, and from pharmaceutical/biological companies: Baxter, Bayer Schering, Biogen Idec, CSL, Genzyme, Grifols, Octapharma, Merck, Novartis, Roche, Sanofi Aventis Genzyme, Servier, and TEVA. Relevant to this study, Biogen is the manufacturer of natalizumab. S.W.R. is a shareholder of Medical Safety Systems trading as RxMx (grant and contracts with Genzyme >$25,000 AUD, contracts with Novartis, Roche, Janssen). M.R. received speaker honoraria from Novartis, Bayer Vital GmbH, and Ipsen and travel reimbursement from Bayer Schering, Biogen Idec, Merz, Genzyme, Teva, and Merck, none related to this study. J.D. received research support by Bayer and Novartis, travel support by Bayer, Novartis, Biogen, and Merck Serono, and honoraria for lectures and advisory by Bayer, Novartis, Biogen, Merck Serono, Roche, and Sanofi Genzyme. B.W. received research support from German Ministry of Education and Research, Dietmar Hopp Foundation, Klaus Tschira Foundation, Sanofi Genzyme, Merck Serono, and Novartis and speaker honoraria and/or travel support from Bayer Healthcare, Biogen, Merck Serono, Novartis, Sanofi Genzyme, and TEVA outside the submitted work. M.K. received travel support and honoraria from Bayer Schering, Biogen Idec, Chugai Pharma, Merck Serono, Novartis Pharma, Teva Pharma, and Shire Deutschland. H.L. received honoraria for lectures and consultation related to multiple sclerosis from Novartis, Roche, Sanofi Aventis, Biogen, and MEDDAY. R.H. received speaker honoraria from Euroimmun and research support from the Jubiläumsfonds der Östereichischen Nationalbank (project 16919) and the Austrian Science Fund (FWF: I3334-B27). N.S. received travel support from Novartis and Sanofi-Genzyme. L.K. received compensation for serving on scientific advisory boards for Genzyme, Merck, Novartis, and Roche; speaker honoraria and travel support from Biogen, Genzyme, Merck, Novartis, and Roche; and research support from Biogen, Genzyme, Merck, Novartis, and Roche, the German Ministry for Education and Research (BMBF), Deutsche Forschungsgesellschaft (DFG), and the Interdisciplinary Center for Clinical Studies (IZKF) Münster. S.G.M. receives honoraria for lecturing, and travel expenses for attending meetings from Almirall, Amicus Therapeutics Germany, Bayer Health Care, Biogen, Celgene, Diamed, Genzyme, MedDay Pharmaceuticals, Merck Serono, Novartis, Novo Nordisk, ONO Pharma, Roche, Sanofi-Aventis, Chugai Pharma, QuintilesIMS, and Teva. His research is funded by the German Ministry for Education and Research (BMBF), Deutsche Forschungsgesellschaft (DFG), Else Kröner Fresenius Foundation, German Academic Exchange Service, Hertie Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Muenster, German Foundation Neurology and Almirall, Amicus Therapeutics Germany, Biogen, Diamed, Fresenius Medical Care, Genzyme, Merck Serono, Novartis, ONO Pharma, Roche, and Teva. G.M.-B. received speaker honoraria and travel support for attending meetings from Abbvie, Genzyme, Gilead, and Pfizer. H.W. received honoraria for scientific advisory boards/steering committees from Biogen, Evgen, MedDay Pharmaceuticals, Merck Serono, Novartis, Roche Pharma AG, and Sanofi-Genzyme. He received speaker honoraria and travel support for attending meetings from Alexion, Biogen, Cognomed, F. Hoffmann-La Roche Ltd., Gemeinnützige Hertie-Stiftung, Merck Serono, Novartis, Roche Pharma AG, Sanofi-Genzyme, TEVA, and WebMD Globa. H.W. received compensation as a consultant from Abbvie, Actelion, Biogen, IGES, Novartis, Roche, Sanofi-Genzyme, and the Swiss Multiple Sclerosis Society. He also received research support from the German Ministry for Education and Research (BMBF), Deutsche Forschungsgesellschaft (DFG), Else Kröner Fresenius Foundation, Fresenius Foundation, Hertie Foundation, NRW Ministry of Education and Research, Interdisciplinary Center for Clinical Studies (IZKF) Muenster and RE Children’s Foundation, Biogen GmbH, GlaxoSmithKline GmbH, Roche Pharma AG, and Sanofi-Genzyme. R.L. received grant support from Pierre Fabre, GlaxoSmithKline, and Diaccurate. He received speaker or scientific board honoraria from Biogen, Servier, Novartis, and Sanofi-Genzyme. R.L. is currently receiving grants from GlaxoSmithKline, Cancer Research Institute, French Cancer research foundation (ARC), Rare Diseases Foundation, and National Institute of Cancer (INCa). C.M., U.B., L.Y., A.T., S.H., T.S.-H., H.P., M.S., D.L., M.E.B., E.B., and K.D. have no financial disclosures.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
