

Effector-triggered immunity and pathogen sensing in metazoans
- PMID: 31857733
- PMCID: PMC9326894
- DOI: 10.1038/s41564-019-0623-2
Effector-triggered immunity and pathogen sensing in metazoans
Erratum in
-
Publisher Correction: Effector-triggered immunity and pathogen sensing in metazoans.Nat Microbiol. 2020 Mar;5(3):528. doi: 10.1038/s41564-020-0682-4. Nat Microbiol. 2020. PMID: 32042131 Free PMC article.
Abstract
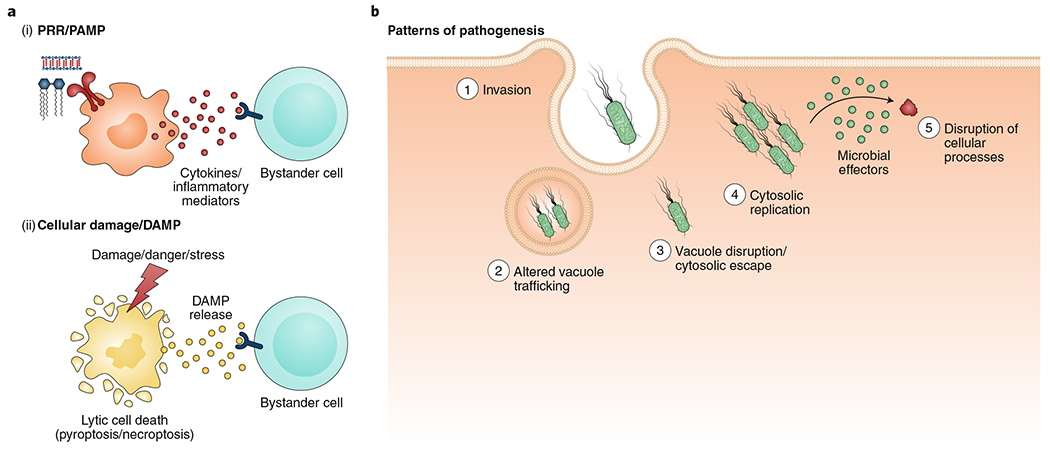
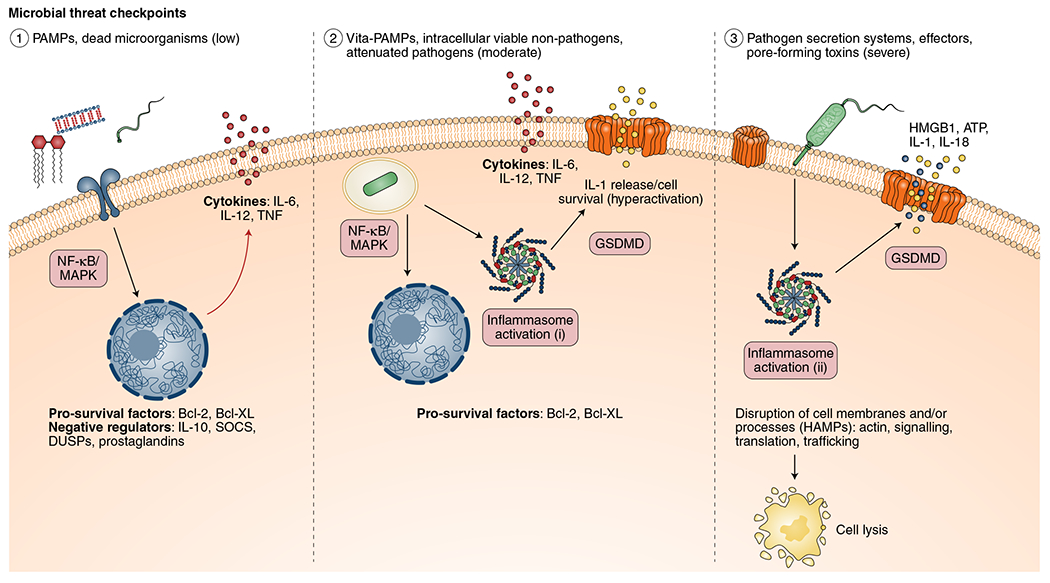
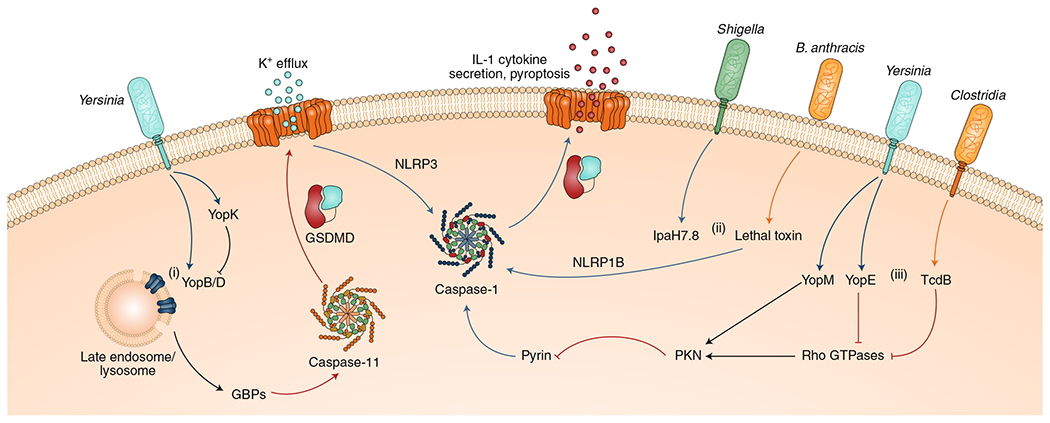
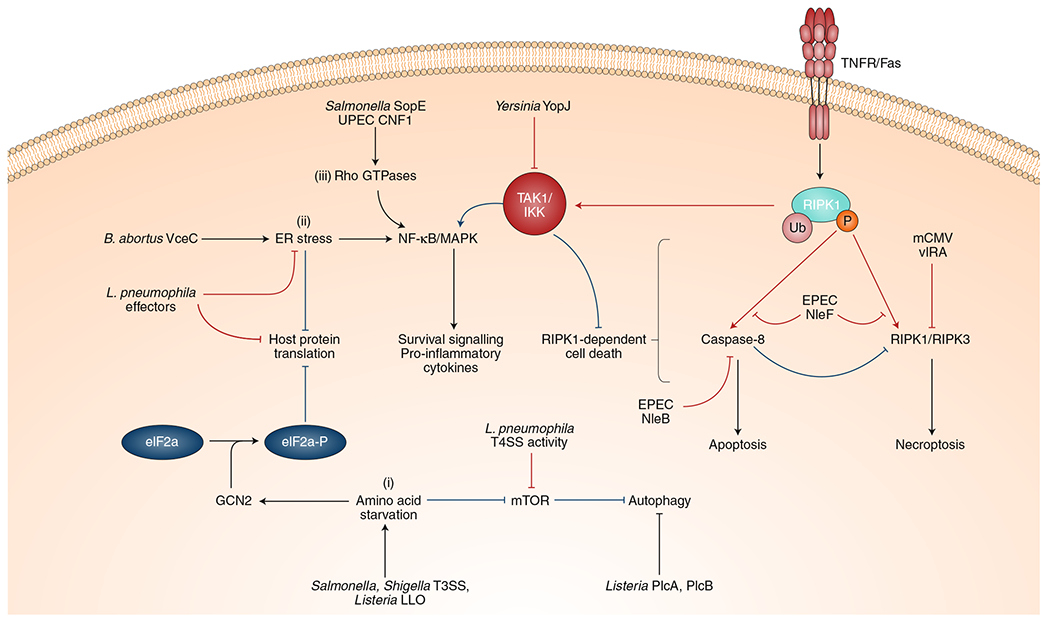
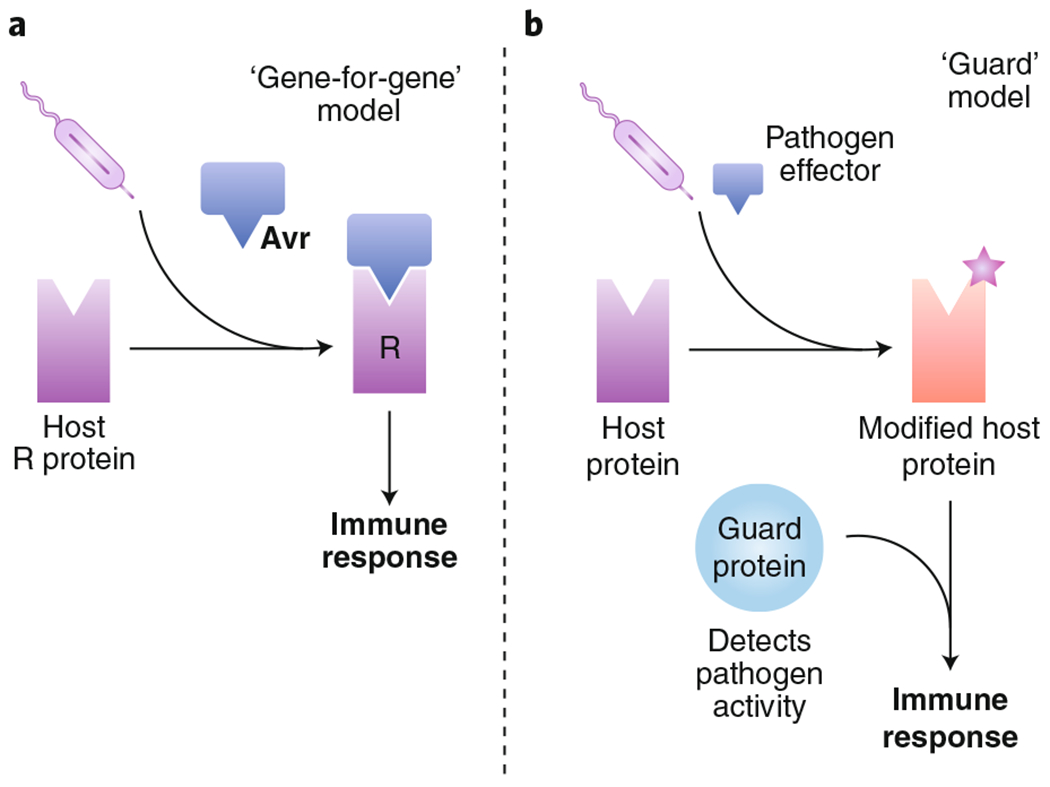
Microbial pathogens possess an arsenal of strategies to invade their hosts, evade immune defences and promote infection. In particular, bacteria use virulence factors, such as secreted toxins and effector proteins, to manipulate host cellular processes and establish a replicative niche. Survival of eukaryotic organisms in the face of such challenge requires host mechanisms to detect and counteract these pathogen-specific virulence strategies. In this Review, we focus on effector-triggered immunity (ETI) in metazoan organisms as a mechanism for pathogen sensing and distinguishing pathogenic from non-pathogenic microorganisms. For the purposes of this Review, we adopt the concept of ETI formulated originally in the context of plant pathogens and their hosts, wherein specific host proteins 'guard' central cellular processes and trigger inflammatory responses following pathogen-driven disruption of these processes. While molecular mechanisms of ETI are well-described in plants, our understanding of functionally analogous mechanisms in metazoans is still emerging. In this Review, we present an overview of ETI in metazoans and discuss recently described cellular processes that are guarded by the host. Although all pathogens manipulate host pathways, we focus primarily on bacterial pathogens and highlight pathways of effector-triggered immune defence that sense disruption of core cellular processes by pathogens. Finally, we discuss recent developments in our understanding of how pathogens can evade ETI to overcome these host adaptations.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Janeway CA Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol 54, 1–13 (1989). - PubMed
-
- Ausubel FM Are innate immune signaling pathways in plants and animals conserved? Nat. Immunol 6, 973 (2005). - PubMed
-
- Janeway CA & Medzhitov R Innate immune recognition. Annu. Rev. Immunol 20, 197–216 (2002). - PubMed
-
- Pasare C & Medzhitov R Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299, 1033–1036 (2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01AI139102A1/U.S. Department of Health & Human Services | NIH | National Institute of Allergy and Infectious Diseases (NIAID)/International
- R01 AI103062/AI/NIAID NIH HHS/United States
- R21 AI135421/AI/NIAID NIH HHS/United States
- T32 GM007229/GM/NIGMS NIH HHS/United States
- R01 AI128530/AI/NIAID NIH HHS/United States
- R01 AI123243/AI/NIAID NIH HHS/United States
- R01AI118861/U.S. Department of Health & Human Services | NIH | National Institute of Allergy and Infectious Diseases (NIAID)/International
- R01 AI118861/AI/NIAID NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01AI123243/U.S. Department of Health & Human Services | NIH | National Institute of Allergy and Infectious Diseases (NIAID)/International
- R21AI135421/U.S. Department of Health & Human Services | NIH | National Institute of Allergy and Infectious Diseases (NIAID)/International
- R01 AI139102/AI/NIAID NIH HHS/United States
- R01AI128530/U.S. Department of Health & Human Services | NIH | National Institute of Allergy and Infectious Diseases (NIAID)/International
LinkOut - more resources
Full Text Sources
Medical
