

Pulmonary arterial hypertension populations of special interest: portopulmonary hypertension and pulmonary arterial hypertension associated with congenital heart disease
- PMID: 31857799
- PMCID: PMC6915053
- DOI: 10.1093/eurheartj/suz221
Pulmonary arterial hypertension populations of special interest: portopulmonary hypertension and pulmonary arterial hypertension associated with congenital heart disease
Abstract
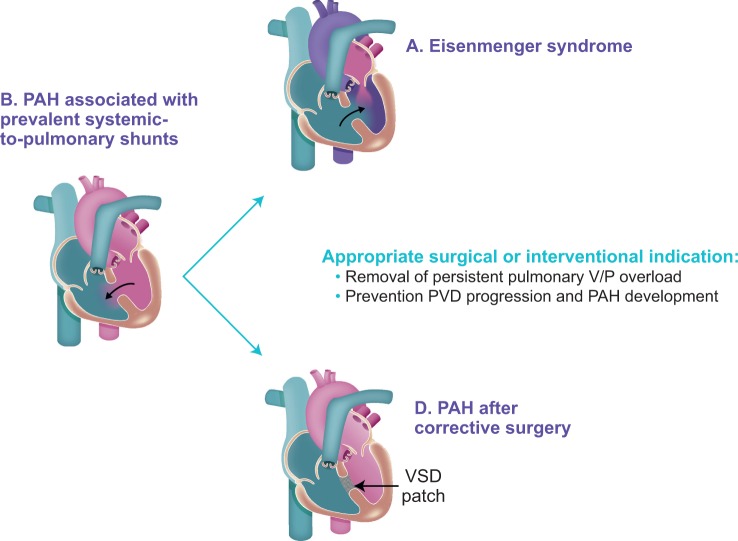
Guidelines exist for management of pulmonary arterial hypertension (PAH), but information is limited for certain patient subgroups, including adults with portopulmonary hypertension (PoPH) or with PAH associated with congenital heart disease (PAH-CHD). This article discusses screening, clinical management, and prognosis in PoPH and PAH-CHD and, as such, considers the most recent clinical data and expert advice. A multidisciplinary consultation and follow-up by specialists are crucial for management of both PoPH and PAH-CHD, but each condition presents with unique challenges. Development of PoPH most commonly occurs among patients with liver cirrhosis. Initially, patients may be asymptomatic for PoPH and, if untreated, survival with PoPH is generally worse than with idiopathic PAH (IPAH), so early identification with screening is crucial. PoPH can be managed with PAH-specific pharmacological therapy, and resolution is possible in some patients with liver transplantation. With PAH-CHD, survival rates are typically higher than with IPAH but vary across the four subtypes: Eisenmenger syndrome, systemic-to-pulmonary shunts, small cardiac defects, and corrected defects. Screening is also crucial and, in patients who undergo correction of CHD, the presence of PAH should be assessed immediately after repair and throughout their long-term follow-up, with frequency of assessments determined by the patient's characteristics at the time of correction. Early screening for PAH in patients with portal hypertension or CHD, and multidisciplinary management of PoPH or PAH-CHD are important for the best patient outcomes.
Keywords: Congenital heart disease; Liver transplant; Portopulmonary hypertension; Pulmonary arterial hypertension.
Published on behalf of the European Society of Cardiology. © The Author(s) 2019.
Figures

References
-
- Galiè N, Humbert M, Vachiéry JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M; ESC Scientific Document Group. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J 2016;37:67–119. - PubMed
-
- Galiè N, Beghetti M, Gatzoulis MA, Granton J, Berger RM, Lauer A, Chiossi E, Landzberg M.. Bosentan randomized trial of endothelin antagonist therapy I. Bosentan therapy in patients with Eisenmenger syndrome: a multicenter, double-blind, randomized, placebo-controlled study. Circulation 2006;114:48–54. - PubMed
-
- Sitbon O, Bosch J, Cottreel E, Csonka D, de Groote P, Hoeper MM, Kim NH, Martin N, Savale L, Krowka M.. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir Med 2019;7:594–604. - PubMed
-
- Dardi F, Manes A, Palazzini M, Bachetti C, Mazzanti G, Rinaldi A, Albini A, Gotti E, Monti E, Bacchi Reggiani ML, Galiè N.. Combining bosentan and sildenafil in pulmonary arterial hypertension patients failing monotherapy: real-world insights. Eur Respir J 2015;46:414–421. - PubMed