

Crossover interference and sex-specific genetic maps shape identical by descent sharing in close relatives
- PMID: 31860654
- PMCID: PMC6944377
- DOI: 10.1371/journal.pgen.1007979
Crossover interference and sex-specific genetic maps shape identical by descent sharing in close relatives
Abstract
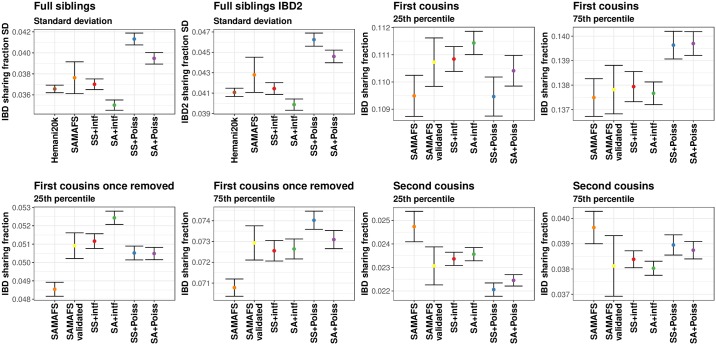
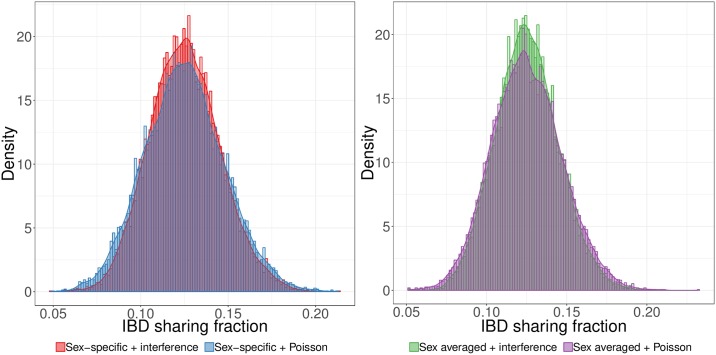
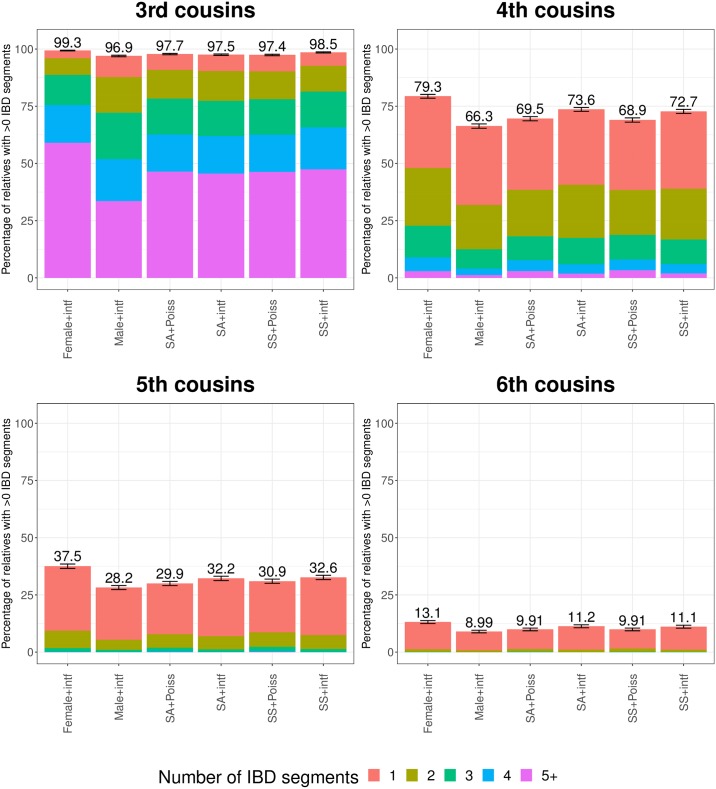
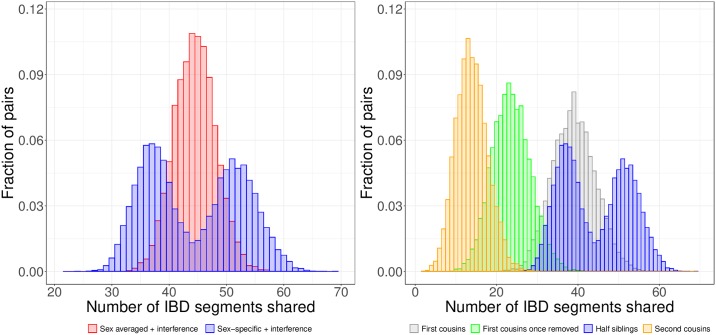
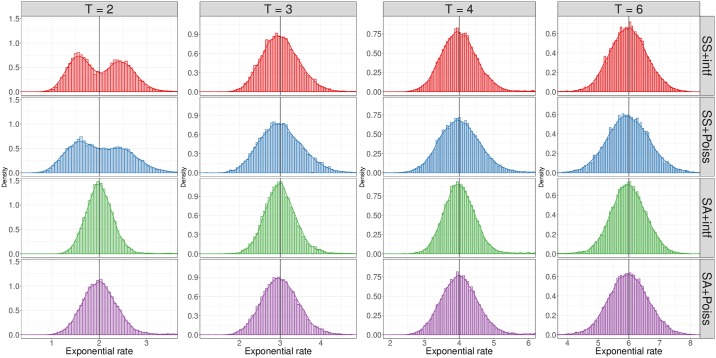
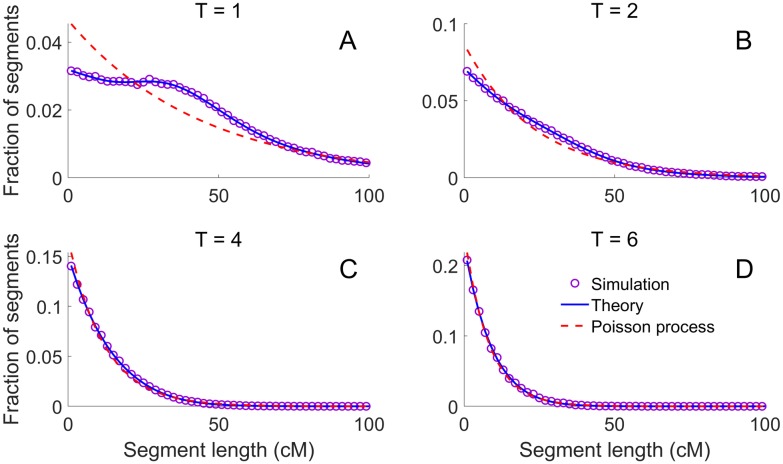
Simulations of close relatives and identical by descent (IBD) segments are common in genetic studies, yet most past efforts have utilized sex averaged genetic maps and ignored crossover interference, thus omitting features known to affect the breakpoints of IBD segments. We developed Ped-sim, a method for simulating relatives that can utilize either sex-specific or sex averaged genetic maps and also either a model of crossover interference or the traditional Poisson model for inter-crossover distances. To characterize the impact of previously ignored mechanisms, we simulated data for all four combinations of these factors. We found that modeling crossover interference decreases the standard deviation of pairwise IBD proportions by 10.4% on average in full siblings through second cousins. By contrast, sex-specific maps increase this standard deviation by 4.2% on average, and also impact the number of segments relatives share. Most notably, using sex-specific maps, the number of segments half-siblings share is bimodal; and when combined with interference modeling, the probability that sixth cousins have non-zero IBD sharing ranges from 9.0 to 13.1%, depending on the sexes of the individuals through which they are related. We present new analytical results for the distributions of IBD segments under these models and show they match results from simulations. Finally, we compared IBD sharing rates between simulated and real relatives and find that the combination of sex-specific maps and interference modeling most accurately captures IBD rates in real data. Ped-sim is open source and available from https://github.com/williamslab/ped-sim.
Conflict of interest statement
Shai Carmi is a paid consultant for MyHeritage. All other authors declare no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
