

From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data
- PMID: 31861212
- PMCID: PMC6950334
- DOI: 10.3390/metabo9120308
From Samples to Insights into Metabolism: Uncovering Biologically Relevant Information in LC-HRMS Metabolomics Data
Abstract
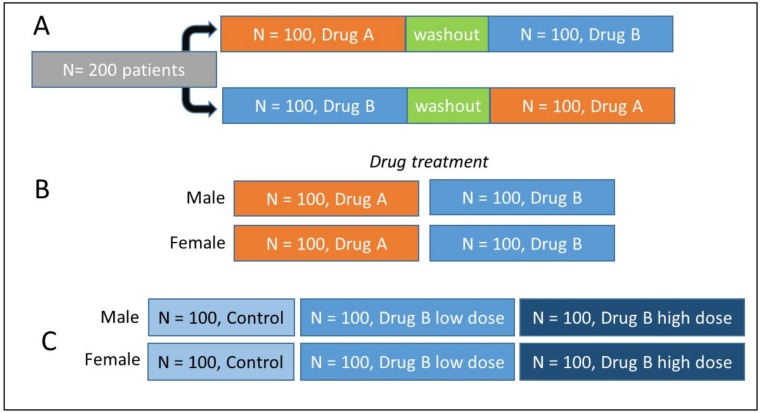
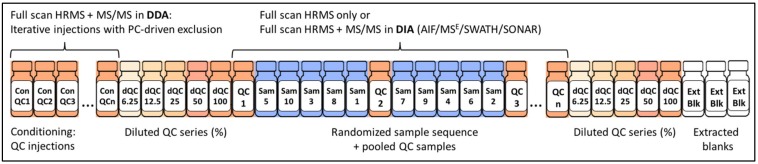
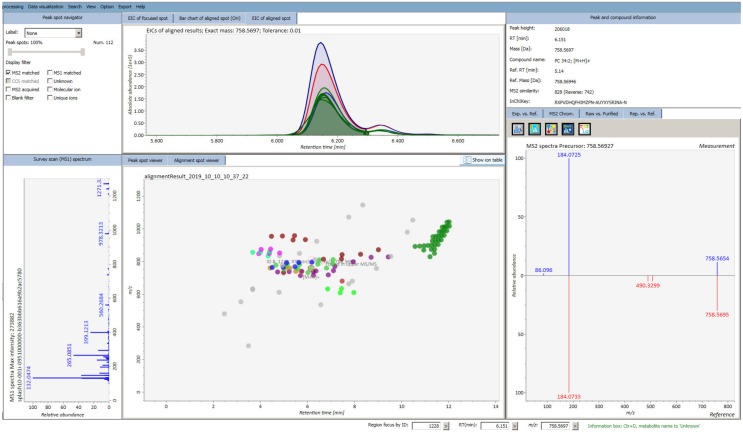
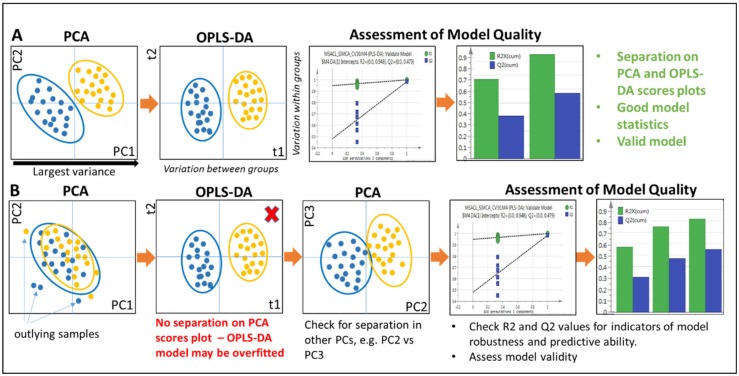
Untargeted metabolomics (including lipidomics) is a holistic approach to biomarker discovery and mechanistic insights into disease onset and progression, and response to intervention. Each step of the analytical and statistical pipeline is crucial for the generation of high-quality, robust data. Metabolite identification remains the bottleneck in these studies; therefore, confidence in the data produced is paramount in order to maximize the biological output. Here, we outline the key steps of the metabolomics workflow and provide details on important parameters and considerations. Studies should be designed carefully to ensure appropriate statistical power and adequate controls. Subsequent sample handling and preparation should avoid the introduction of bias, which can significantly affect downstream data interpretation. It is not possible to cover the entire metabolome with a single platform; therefore, the analytical platform should reflect the biological sample under investigation and the question(s) under consideration. The large, complex datasets produced need to be pre-processed in order to extract meaningful information. Finally, the most time-consuming steps are metabolite identification, as well as metabolic pathway and network analysis. Here we discuss some widely used tools and the pitfalls of each step of the workflow, with the ultimate aim of guiding the reader towards the most efficient pipeline for their metabolomics studies.
Keywords: data processing; experimental design; liquid chromatography–mass spectrometry (LC-MS); metabolic pathway and network analysis; metabolism; metabolite identification; sample preparation; univariate and multivariate statistics; untargeted metabolomics.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Chong J., Wishart D.S., Xia J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinformatics. 2019;68:e86. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
