

Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets
- PMID: 31862961
- PMCID: PMC6925302
- DOI: 10.1038/s41467-019-13805-y
Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets
Abstract
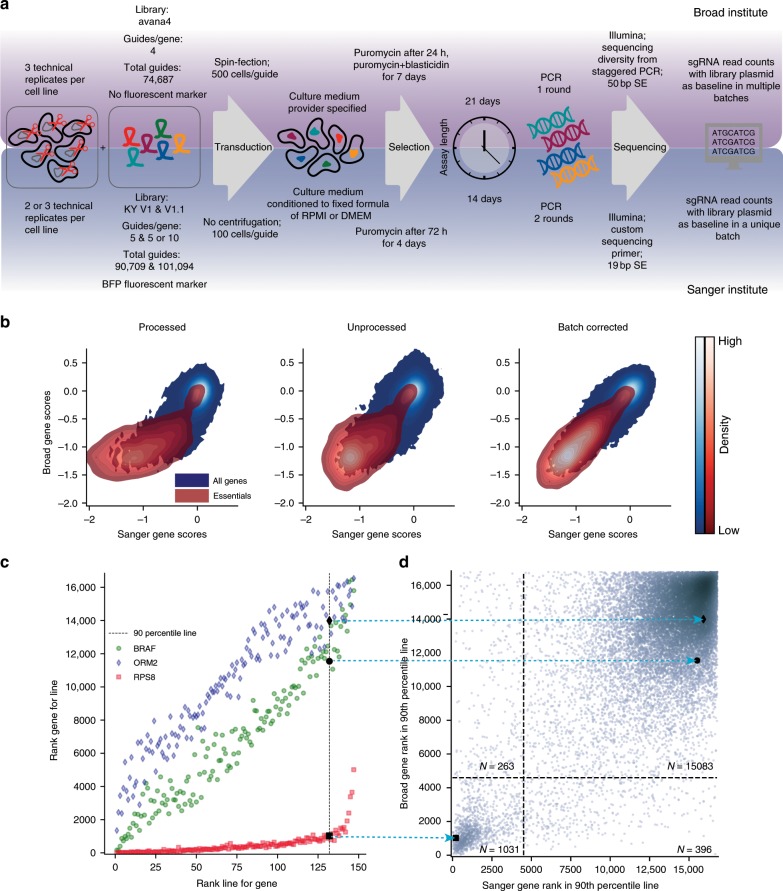
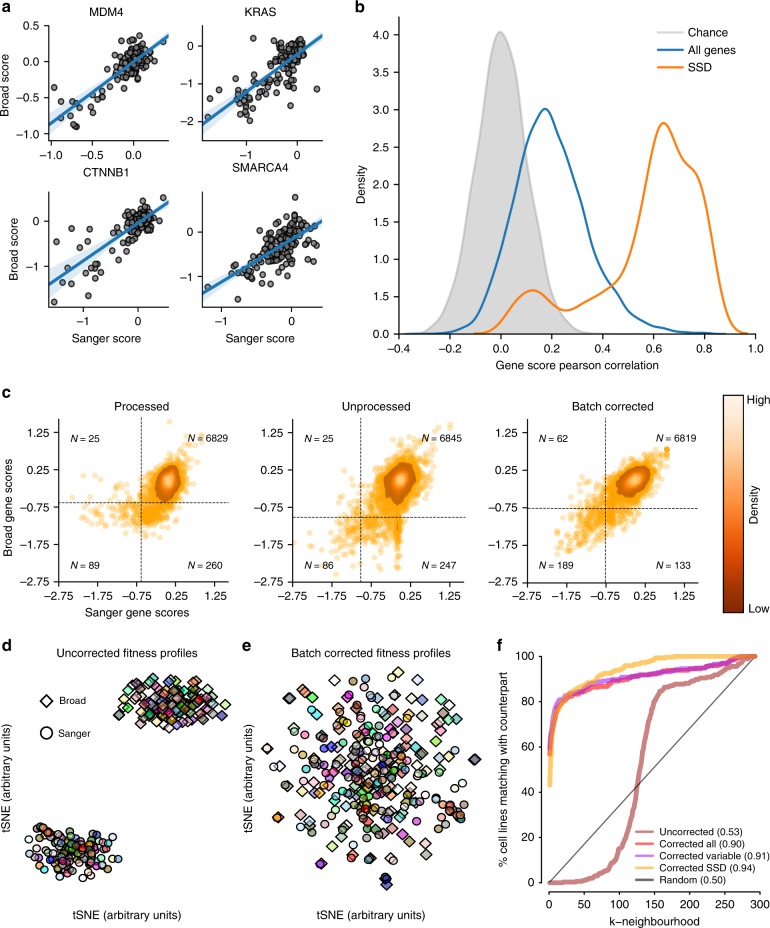
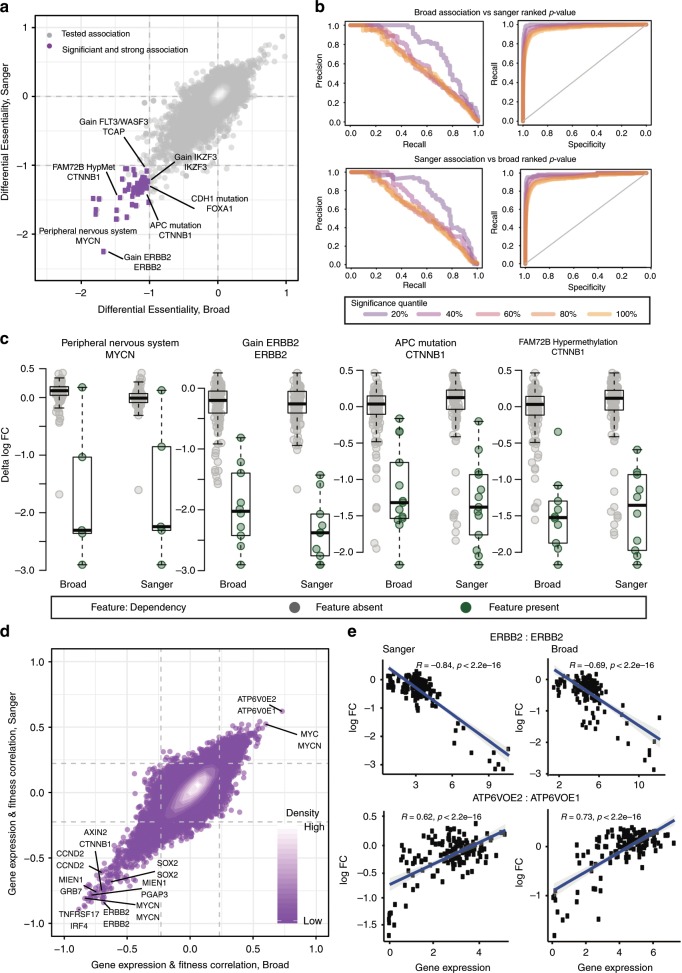
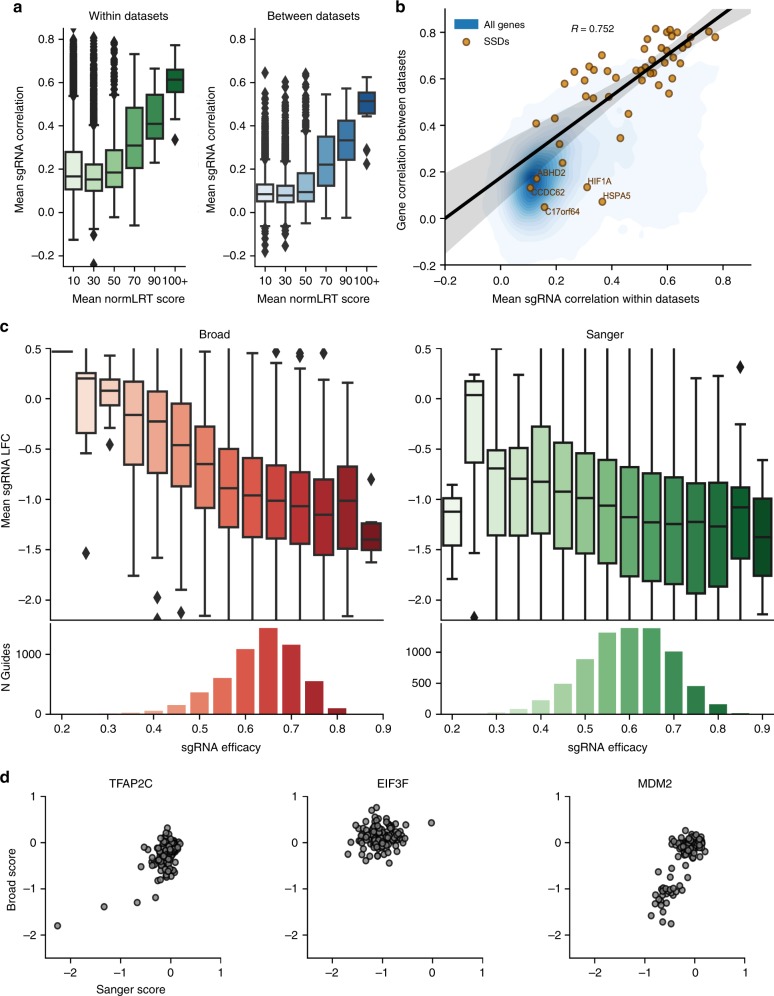
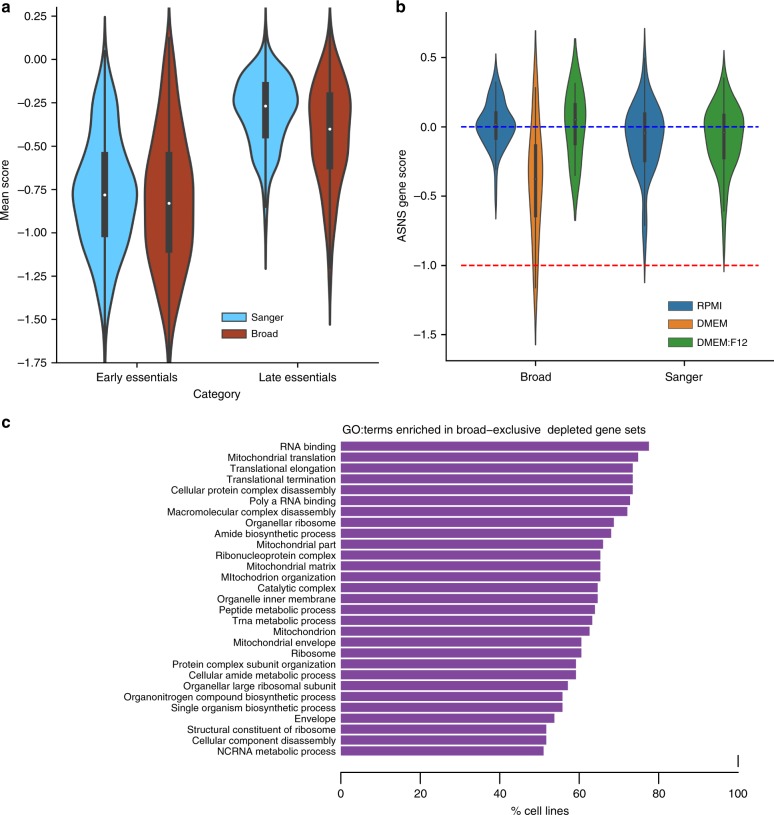
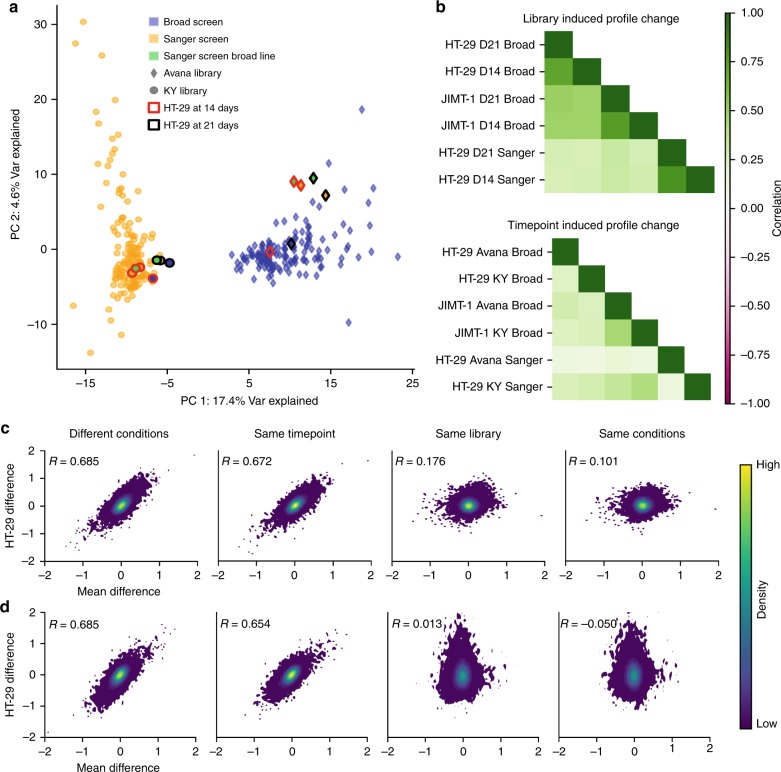
Genome-scale CRISPR-Cas9 viability screens performed in cancer cell lines provide a systematic approach to identify cancer dependencies and new therapeutic targets. As multiple large-scale screens become available, a formal assessment of the reproducibility of these experiments becomes necessary. We analyze data from recently published pan-cancer CRISPR-Cas9 screens performed at the Broad and Sanger Institutes. Despite significant differences in experimental protocols and reagents, we find that the screen results are highly concordant across multiple metrics with both common and specific dependencies jointly identified across the two studies. Furthermore, robust biomarkers of gene dependency found in one data set are recovered in the other. Through further analysis and replication experiments at each institute, we show that batch effects are driven principally by two key experimental parameters: the reagent library and the assay length. These results indicate that the Broad and Sanger CRISPR-Cas9 viability screens yield robust and reproducible findings.
Conflict of interest statement
C.P., F.M.B., H.N., M.J.G. and F.I. receive funding from Open Targets, a public-private initiative involving academia and industry. K.Y. and M.J.G. receive funding from AstraZeneca. M.J.G performed consultancy for Sanofi. J.G.D. and A.T. perform consulting for Tango Therapeutics. W.C.H. performs consulting for Thermo Fisher, AdjulB, MBM Capital, and Paraxel, and is a founder and scientific advisory board member of KSQ Therapeutics. T.R.G. performs consulting for GlaxoSmithKline, Sherlock Biosciences, and Foundation Medicine. F.I. performs consultancy for the joint CRUK - AstraZeneca Functional Genomics Centre. All the other authors declare no competing interests.
Figures
Comment in
-
Cancer Screens: Better Together.CRISPR J. 2020 Feb;3(1):12-14. doi: 10.1089/crispr.2020.29084.spm. CRISPR J. 2020. PMID: 32091251 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
